Using association rule mining and ontologies to generate metadata recommendations from multiple biomedical databases¶
Marcos Martínez-Romero*, Martin J. O'Connor, Attila L. Egyedi, Debra Willrett, Josef Hardi, John Graybeal, and Mark A. Musen
Stanford Center for Biomedical Informatics Research, 1265 Welch Road, Stanford University School of Medicine, Stanford, CA 94305-5479, USA
* Correspondence: marcosmr@stanford.edu
Purpose of this document¶
This document is a Jupyter notebook that describes how to reproduce the evaluation presented in our paper. The notebook uses mostly Python but includes some references to R scripts used to generate the paper figures.
The scripts used to generate the results and figures in the paper are in the scripts folder. The results generated when running the code cells in this notebook will be saved to a local workspace folder.
Table of contents¶
Viewing and running this notebook¶
GitHub will automatically generate a static online view of this notebook. However, current GitHub's rendering does not support some features, such as the anchor links that connect the 'Table of contents' to the different sections. A more reliable way to view the notebook file online is by using nbviewer, which is the official viewer of the Jupyter Notebook project. Click here to open our notebook using nbviewer.
The interactive features of our notebook will not work neither from GitHub nor nbviewer. For a fully interactive version of this notebook, you can set up a Jupyter Notebook server locally and start it from the local folder where you cloned the repository. For more information, see Jupyter's official documentation. Once your local Jupyter Notebook server is running, go to http://localhost:8888/ and click on ValueRecommenderEvaluation.ipynb to open our notebook. You can also run the notebook on Binder by clicking here.
Download data.zip¶
Run the following code cell to download a data.zip file (1.57GB) that contains all the input data used in our evaluation. The file contains a data folder with the inputs and outputs of the different evaluation steps, including the rules used to train the system, as well as the results of all the experiments. Alternatively, you can download and unzip the file manually using this link.
IMPORTANT: Some of the links in the rest of the notebook will not work until the data.zip file has been downloaded and extracted to a local data folder.
# Download data.zip file from Google Drive and unzip it to a local 'data' folder
from google_drive_downloader import GoogleDriveDownloader as gdd
gdd.download_file_from_google_drive(file_id='1AjcgMi3VM1sYdshAkcUiYhP4QBhqhtfk',
dest_path='./data.zip',
unzip=True, overwrite=True)
Downloading 1AjcgMi3VM1sYdshAkcUiYhP4QBhqhtfk into ./data.zip... Done. Unzipping...Done.
Step 1: Datasets download¶
1.a. NCBI BioSample¶
We downloaded the full content of the NCBI BioSample database from the NCBI BioSample FTP repository as a .gz file, which you can find in the data/samples/ncbi_samples/original folder. This file contains metadata about 7.8M NCBI samples. To begin, copy the file to the workspace folder:
%%time
# Copy the .gz file with the NCBI samples to your workspace folder
from shutil import copy
import os
import scripts.constants as c
source_file_path = c.NCBI_SAMPLES_ORIGINAL_FILE_PATH
dest_path = os.path.join(c.WORKSPACE_FOLDER, c.NCBI_SAMPLES_ORIGINAL_PATH)
print('Source file path: ' + source_file_path)
print('Destination path: ' + dest_path)
dest_file_name = c.NCBI_SAMPLES_FILE_DEST
if not os.path.exists(dest_path):
os.makedirs(dest_path)
copy(c.NCBI_SAMPLES_ORIGINAL_FILE_PATH, os.path.join(dest_path, dest_file_name))
Source file path: data/samples/ncbi_samples/original/2018-03-09-biosample_set.xml.gz Destination path: workspace/data/samples/ncbi_samples/original CPU times: user 288 ms, sys: 1.09 s, total: 1.38 s Wall time: 2.11 s
Alternative: Note that the NCBI samples file was downloaded on March 9, 2018. Alternatively, if you want to conduct the evaluation with the most recent NCBI samples, run the following cell:
# OPTIONAL: Download the most recent NCBI biosamples to the workspace
import zipfile
import urllib.request
import sys
import os
import time
import scripts.util as util
import scripts.constants as c
url = c.NCBI_DOWNLOAD_URL
dest_path = os.path.join(c.WORKSPACE_FOLDER, c.NCBI_SAMPLES_ORIGINAL_PATH)
dest_file_name = c.NCBI_SAMPLES_FILE_DEST
print('Source URL: ' + url)
print('Destination path: ' + dest_path)
if not os.path.exists(dest_path):
os.makedirs(dest_path)
urllib.request.urlretrieve(url, os.path.join(dest_path, dest_file_name), reporthook=util.log_progress)
1.b. EBI BioSamples¶
We wrote a script (ebi_biosamples_1_download_split.py) to download all samples metadata from the EBI BioSamples database using the EBI BioSamples API. We stored the results as a ZIP file 2018-03-09-ebi_samples.zip that contains 412 JSON files with metadata for 4.1M samples in total. Extract the file to the workspace:
import zipfile, os
import scripts.constants as c
source_path = c.EBI_SAMPLES_ORIGINAL_FILE_PATH
dest_path = os.path.join(c.WORKSPACE_FOLDER, c.EBI_SAMPLES_ORIGINAL_PATH)
with zipfile.ZipFile(c.EBI_SAMPLES_ORIGINAL_FILE_PATH, 'r') as zip_obj:
zip_obj.extractall(dest_path)
Alternative: Note that these EBI samples were downloaded on March 9, 2018. If you want to run the evaluation with the most recent EBI samples, you can run ebi_biosamples_1_download_split.py again:
# Alternative: download all the EBI samples from the EBI's API
%run ./scripts/ebi_biosamples_1_download_split
Step 2: Generation of template instances¶
2.1. Determine relevant attributes and create CEDAR templates¶
2.1.a. NCBI BioSample¶
For NCBI BioSample, we created a CEDAR template with all the attributes defined by the NCBI BioSample Human Package v1.0, which are: biosample_accession, sample_name, sample_title, bioproject_accession, organism, isolate, age, biomaterial_provider, sex, tissue, cell_line, cell_subtype, cell_type, culture_collection, dev_stage, disease, disease_stage, ethnicity, health_state, karyotype, phenotype, population, race, sample_type, treatment, description. The template is available here.
2.1.b. EBI BioSamples¶
The EBI BioSamples API's output format defines some top-level attributes and makes it possible to add new attributes that describe sample characteristics:
{
"accession": "...",
"name": "...",
"releaseDate": "...",
"updateDate": "...",
"characteristics": { // key-value pairs (e.g., organism, age, sex, etc.)
...
},
"organization": "...",
"contact": "..."
}
Based on this format, we defined a metadata template containing 14 fields with general metadata about biological samples and some additional fields that capture specific characteristics of human samples: accession, name, releaseDate, updateDate, organization, contact, organism, age, sex, organismPart, cellLine, cellType, diseaseState, ethnicity. The template is available here.
We focused our analysis on the subset of fields that meet two key requirements: (1) they are present in both templates and, therefore, can be used to evaluate cross-template recommendations; and (2) they contain categorical values, that is, they represent information about discrete characteristics. We selected 6 fields that met these criteria. These fields are: sex, organism part, cell line, cell type, disease, and ethnicity. The names used to refer to these fields in both CEDAR's NCBI BioSample template and CEDAR's EBI BioSamples template are shown in the following table:
| Characteristic | NCBI BioSample attribute name | EBI BioSamples attribute name |
|---|---|---|
| sex | sex | sex |
| organism part | tissue | organismPart |
| cell line | cell_line | cellLine |
| cell type | cell_type | cellType |
| disease | disease | diseaseState |
| ethnicity | ethnicity | ethnicity |
2.2. Select samples¶
We filtered the samples based on two criteria:
- The sample is from "Homo sapiens" (organism=Homo sapiens).
- The sample has non-empty values for at least 3 of the 6 fields in the previous table.
2.2.a. NCBI BioSample¶
Script used: ncbi_biosample_1_filter.py.
# Filter the NCBI samples
%run ./scripts/ncbi_biosample_1_filter.py
The result is an XML file with 157,653 samples (biosample_result_filtered.xml).
Shortcut: Copy the precomputed NCBI filtered samples to the workspace:
# Shortcut: reuse existing filtered NCBI samples
import os
from shutil import copyfile
import scripts.arm_constants as c
src = c.NCBI_FILTER_OUTPUT_FILE_PRECOMPUTED
dst = c.NCBI_FILTER_OUTPUT_FILE
if not os.path.exists(os.path.dirname(dst)):
os.makedirs(os.path.dirname(dst))
copyfile(src, dst)
'./workspace/data/samples/ncbi_samples/filtered/biosample_result_filtered.xml'
2.2.b. EBI BioSamples¶
In the case of the EBI samples, we used the script ebi_biosamples_2_filter.py
# Filter the EBI samples
%run ./scripts/ebi_biosamples_2_filter.py
Results: 14 JSON files with a total of 135,187 samples, which are available in this folder.
Shortcut: Copy the precomputed EBI filtered samples to the workspace:
# Shortcut: reuse existing filtered EBI samples
import os
from shutil import copyfile
import scripts.arm_constants as c
src = c.EBI_FILTER_OUTPUT_FOLDER_PRECOMPUTED
dst = c.EBI_FILTER_OUTPUT_FOLDER
if not os.path.exists(dst):
os.makedirs(dst)
for file_name in os.listdir(src):
print(file_name)
print(os.path.join(src, file_name))
print(os.path.join(dst, file_name))
copyfile(os.path.join(src, file_name), os.path.join(dst, file_name))
ebi_biosamples_filtered_3_20000to29999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_3_20000to29999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_3_20000to29999.json ebi_biosamples_filtered_1_0to9999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_1_0to9999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_1_0to9999.json ebi_biosamples_filtered_2_10000to19999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_2_10000to19999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_2_10000to19999.json ebi_biosamples_filtered_4_30000to39999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_4_30000to39999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_4_30000to39999.json ebi_biosamples_filtered_10_90000to99999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_10_90000to99999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_10_90000to99999.json ebi_biosamples_filtered_5_40000to49999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_5_40000to49999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_5_40000to49999.json ebi_biosamples_filtered_6_50000to59999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_6_50000to59999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_6_50000to59999.json ebi_biosamples_filtered_7_60000to69999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_7_60000to69999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_7_60000to69999.json ebi_biosamples_filtered_9_80000to89999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_9_80000to89999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_9_80000to89999.json ebi_biosamples_filtered_8_70000to79999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_8_70000to79999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_8_70000to79999.json ebi_biosamples_filtered_13_120000to129999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_13_120000to129999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_13_120000to129999.json ebi_biosamples_filtered_11_100000to109999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_11_100000to109999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_11_100000to109999.json ebi_biosamples_filtered_14_130000to135186.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_14_130000to135186.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_14_130000to135186.json ebi_biosamples_filtered_12_110000to119999.json ./data/samples/ebi_samples/filtered/ebi_biosamples_filtered_12_110000to119999.json ./workspace/data/samples/ebi_samples/filtered/ebi_biosamples_filtered_12_110000to119999.json
2.3. Generate CEDAR instances¶
We transformed the NCBI and EBI samples obtained from the previous step to CEDAR template instances conforming to CEDAR's JSON-based Template Model.
For NCBI samples, we used the script ncbi_biosample_2_to_cedar_instances.py:
%%time
# Generate CEDAR instances from NCBI samples
%run ./scripts/ncbi_biosample_2_to_cedar_instances.py
Reading file: ./workspace/data/samples/ncbi_samples/filtered/biosample_result_filtered.xml Extracting all samples from file (no. samples: 157653) Randomly picking 135187 samples Generating CEDAR instances... No. instances generated: 10000(7%) No. instances generated: 20000(15%) No. instances generated: 30000(22%) No. instances generated: 40000(30%) No. instances generated: 50000(37%) No. instances generated: 60000(44%) No. instances generated: 70000(52%) No. instances generated: 80000(59%) No. instances generated: 90000(67%) No. instances generated: 100000(74%) No. instances generated: 110000(81%) No. instances generated: 120000(89%) No. instances generated: 130000(96%) Finished CPU times: user 2min 30s, sys: 44.2 s, total: 3min 14s Wall time: 4min 7s
CEDAR's NCBI instances will be saved to workspace/data/cedar_instances/ncbi_cedar_instances.
For EBI samples, we used the script ebi_biosamples_3_to_cedar_instances.py:
%%time
# Generate CEDAR instances from EBI samples
%run ./scripts/ebi_biosamples_3_to_cedar_instances.py
Reading EBI biosamples from folder: ./workspace/data/samples/ebi_samples/filtered Total no. samples: 135187 Generating CEDAR instances... No. instances generated: 10000(7%) No. instances generated: 20000(15%) No. instances generated: 30000(22%) No. instances generated: 40000(30%) No. instances generated: 50000(37%) No. instances generated: 60000(44%) No. instances generated: 70000(52%) No. instances generated: 80000(59%) No. instances generated: 90000(67%) No. instances generated: 100000(74%) No. instances generated: 110000(81%) No. instances generated: 120000(89%) No. instances generated: 130000(96%) Finished CPU times: user 1min 43s, sys: 46.4 s, total: 2min 30s Wall time: 3min 28s
CEDAR's EBI instances will be saved to workspace/data/cedar_instances/ebi_cedar_instances.
All the CEDAR instances using to evaluate the system are available at data/cedar_instances.
Step 3: Semantic annotation¶
We used the NCBO Annotator via the NCBO BioPortal API to automatically annotate a total of 270,374 template instances (135,187 instances for each template).
3.1. Extraction of unique values from CEDAR instances¶
To avoid making multiple calls to the NCBO Annotator API for the same terms, we first extracted all the unique values in the CEDAR instances.
%%time
# Extract unique values from NCBI and EBI instances
%run ./scripts/cedar_annotator/1_unique_values_extractor.py
Extracting unique values from CEDAR instances... No. instances processed: 10000 No. instances processed: 20000 No. instances processed: 30000 No. instances processed: 40000 No. instances processed: 50000 No. instances processed: 60000 No. instances processed: 70000 No. instances processed: 80000 No. instances processed: 90000 No. instances processed: 100000 No. instances processed: 110000 No. instances processed: 120000 No. instances processed: 130000 No. instances processed: 140000 No. instances processed: 150000 No. instances processed: 160000 No. instances processed: 170000 No. instances processed: 180000 No. instances processed: 190000 No. instances processed: 200000 No. instances processed: 210000 No. instances processed: 220000 No. instances processed: 230000 No. instances processed: 240000 No. instances processed: 250000 No. instances processed: 260000 No. instances processed: 270000 No. unique values extracted: 26556
We processed 270,374 instances and obtained 26,556 unique values (see unique_values.txt).
3.2. Annotation of unique values and generation of mappings¶
We invoked the NCBO Annotator for the unique values obtained from the previous step. Additionally, we took advantage of the output provided by the Annotator API to extract all the different term URIs that map to each term in BioPortal and store all these equivalences into a mappings file.
Script used: 2_unique_values_annotator.py
Note that when running the following cell, you will be asked to enter your BioPortal API key. If you don't have one, follow these instructions.
%%time
# Enter your BioPortal API key
bp_api_key = input('Please, enter you BioPortal API key and press Enter:')
# Annotate unique values and generate mappings file
%run ./scripts/cedar_annotator/2_unique_values_annotator.py --bioportal-api-key $bp_api_key
Shortcut: If you don't have access to the NCBO Annotator or you don't want to wait for the annotation process to finish, copy the files with the annotated values to your workspace:
# Shortcut: reuse previously generated annotations for the unique values
import os
from shutil import copyfile
import scripts.cedar_annotator.annotation_constants as c
def my_copy(src, dst):
if not os.path.exists(os.path.dirname(dst)):
os.makedirs(os.path.dirname(dst))
copyfile(src, dst)
print (src + ' copied to ' + dst)
src1 = c.VALUES_ANNOTATION_OUTPUT_FILE_PATH_1_PRECOMPUTED
dst1 = c.VALUES_ANNOTATION_OUTPUT_FILE_PATH_1
src2 = c.VALUES_ANNOTATION_OUTPUT_FILE_PATH_2_PRECOMPUTED
dst2 = c.VALUES_ANNOTATION_OUTPUT_FILE_PATH_2
my_copy(src1, dst1)
my_copy(src2, dst2)
./data/cedar_instances_annotated/unique_values/unique_values_annotated_1.json copied to ./workspace/data/cedar_instances_annotated/unique_values/unique_values_annotated_1.json ./data/cedar_instances_annotated/unique_values/unique_values_annotated_2.json copied to ./workspace/data/cedar_instances_annotated/unique_values/unique_values_annotated_2.json
3.3. Annotation of CEDAR instances¶
This process uses the annotations generated in the previous step to annotate the values of the CEDAR instances without making any additional calls to the BioPortal API. The resulting instances are saved to workspace/data/cedar_instances_annotated.
Script: 3_cedar_instances_annotator.py
%%time
# Generate annotated CEDAR instances
%run ./scripts/cedar_annotator/3_cedar_instances_annotator.py
Processing instances folder: ./workspace/data/cedar_instances/ncbi_cedar_instances/training No. annotated instances: 10000 No. annotated instances: 20000 No. annotated instances: 30000 No. annotated instances: 40000 No. annotated instances: 50000 No. annotated instances: 60000 No. annotated instances: 70000 No. annotated instances: 80000 No. annotated instances: 90000 No. annotated instances: 100000 No. annotated instances: 110000 No. total values: 379789 No. non annotated values: 55518 (15%) Processing instances folder: ./workspace/data/cedar_instances/ncbi_cedar_instances/testing No. annotated instances: 120000 No. annotated instances: 130000 No. total values: 446822 No. non annotated values: 65348 (15%) Processing instances folder: ./workspace/data/cedar_instances/ebi_cedar_instances/training No. annotated instances: 140000 No. annotated instances: 150000 No. annotated instances: 160000 No. annotated instances: 170000 No. annotated instances: 180000 No. annotated instances: 190000 No. annotated instances: 200000 No. annotated instances: 210000 No. annotated instances: 220000 No. annotated instances: 230000 No. annotated instances: 240000 No. annotated instances: 250000 No. total values: 817139 No. non annotated values: 117270 (14%) Processing instances folder: ./workspace/data/cedar_instances/ebi_cedar_instances/testing No. annotated instances: 260000 No. annotated instances: 270000 No. total values: 882498 No. non annotated values: 126554 (14%) CPU times: user 3min 48s, sys: 2min 12s, total: 6min 1s Wall time: 2h 14min 31s
All the CEDAR instances using to evaluate the system (both in plain text and annotated) are available at data/cedar_instances.
Step 4: Generation of experimental data sets¶
When we generated the CEDAR instances (step 2.3) and the annotated CEDAR instances (step 3.3), we partitioned the resulting instances for each database (NCBI, EBI) into two datasets, with 85% of the data for training and the remaining 15% for testing.
Step 5: Training¶
We mined association rules from the training sets to discover the hidden relationships between metadata fields. We extracted the rules using a local installation of the CEDAR Workbench. We set up the Value Recommender service to read the instance files from a local folder by updating the Constants.java file as follows:
READ_INSTANCES_FROM_CEDAR = false // Read training instances from a local folder
// Apriori configuration:
public static final int APRIORI_MAX_NUM_RULES = 1000000;
public static int MIN_SUPPORTING_INSTANCES = 5;
public static final double MIN_CONFIDENCE = 0.3;
You will have to run the rule extraction process four times, once for each training set. Before each execution, update the variable CEDAR_INSTANCES_PATH with the full path of the corresponding training set:
- Text-based values:
- To extract the NCBI rules:
.../workspace/data/cedar_instances_annotated/ncbi_cedar_instances/training - To extract the EBI rules:
.../workspace/data/cedar_instances_annotated/ebi_cedar_instances/training
- To extract the NCBI rules:
- Ontology-based values:
- To extract the NCBI rules:
.../workspace/data/cedar_instances/ncbi_cedar_instances/training - To extract the EBI rules:
.../workspace/data/cedar_instances/ebi_cedar_instances/training
- To extract the NCBI rules:
Internally, CEDAR's Value Recommender uses a WEKA's implementation of the Apriori algorithm with a minimum support of 5 instances and a confidence of 0.3 by default. The final set of rules were indexed using Elasticsearch.
Update those constants, compile the cedar-valuerecommender-server project and start it locally. You can trigger the rule generation process from the command line using the following curl command:
curl --request POST \
--url https://valuerecommender.metadatacenter.orgx/command/generate-rules/<TEMPLATE_ID> \
--header 'authorization: apiKey <CEDAR_ADMIN_API_KEY>' \
--header 'content-type: application/json' \
--data '{}'
where CEDAR_ADMIN_API_KEY is the API key of the cedar-admin user in your local CEDAR system, and TEMPLATE_ID is the local identifier of the template that you want to extract rules for, that is, either the identifier of the NCBI BioSample template or the EBI BioSamples template.
5.1. Rules generated¶
The following table shows the number of rules produced for each training set and type of metadata. It also provides a link to a .zip file with the rules. These files are also available in the data/rules folder.
| Training set DB | Type of metadata | No. rules generated | No. rules after filtering | Rules file |
|---|---|---|---|---|
| NCBI | Text-based | 52,192 | 30,295 | ncbi-text-rules.zip |
| EBI | Text-based | 36,915 | 24,983 | ebi-text-rules.zip |
| NCBI | Ontology-based | 18,223 | 12,400 | ncbi-ont-rules.zip |
| EBI | Ontology-based | 16,838 | 11,932 | ebi-ont-rules.zip |
We extracted the rules from Elasticsearch using elasticdump as follows:
Export the rules and mappings from Elasticsearch to JSON format:
elasticdump --input=http://localhost:9200/cedar-rules --output=./ncbi-text-mappings.json --type=mappingelasticdump --input=http://localhost:9200/cedar-rules --output=./ncbi-text-data.json --type=data
Import the rules and mappings to Elasticsearch:
elasticdump --input=./ncbi-text-mappings.json --output=http://localhost:9200/cedar-rules --type=mappingselasticdump --input=./ncbi-text-data.json --output=http://localhost:9200/cedar-rules --type=data
Step 6: Testing¶
In this step, we used the produced rules to evaluate the performance of CEDAR's Value Recommender when predicting values from the test sets. We conducted 8 experiments to cover all combinations of recommendation scenario (single-template or cross-template) and metadata type (text-based or ontology-based). These experiments are listed in the following table. The table also contains links to the .csv files with the results obtained, which are also available in the folder data/results/main_experiment.
| Experiment | Rules file | Training DB | Testing DB | Type of metadata | Results file (.zip) |
|---|---|---|---|---|---|
| 1 | ncbi-text-rules | NCBI | NCBI | Text-based | download |
| 2 | ncbi-ont-rules | NCBI | NCBI | Ontology-based | download |
| 3 | ebi-text-rules | EBI | EBI | Text-based | download |
| 4 | ebi-ont-rules | EBI | EBI | Ontology-based | download |
| 5 | ncbi-text-rules | NCBI | EBI | Text-based | download |
| 6 | ncbi-ont-rules | NCBI | EBI | Ontology-based | download |
| 7 | ebi-text-rules | EBI | NCBI | Text-based | download |
| 8 | ebi-ont-rules | EBI | NCBI | Ontology-based | download |
In order to reproduce the results, follow these steps for each of the eight experiments:
Reset the
cedar-rulesindex by using the console commandcedarat rules-regenerateIndex.Restore the corresponding set of rules by running:
elasticdump --input=./<RULES_FILE> --output=http://localhost:9200/cedar-rules --type=dataUpdate the following variables in the arm_constants.py file according to the training and testing databases used.
EVALUATION_TRAINING_DB EVALUATION_TESTING_DB EVALUATION_USE_ANNOTATED_VALUESSet
EVALUATION_TRAINING_DBandEVALUATION_TESTING_DBtoBIOSAMPLES_DB.NCBIor toBIOSAMPLES_DB.EBIdepending on the database used.Set
EVALUATION_USE_ANNOTATED_VALUEStoTruewhen the type of metadata is Ontology-based and toFalseotherwise.For example, for the experiment 1, the values of those variables should be:
EVALUATION_TRAINING_DB = BIOSAMPLES_DB.NCBI EVALUATION_TESTING_DB = BIOSAMPLES_DB.NCBI EVALUATION_USE_ANNOTATED_VALUES = FalseAfter completing the steps 1-3 and with the CEDAR Workbench running in your local machine, the evaluation process can be started by executing the script arm_evaluation_main.py with your CEDAR API key.
%%time
# Enter your CEDAR API key
cedar_api_key = input('Please, enter you CEDAR API key and press Enter: ')
# Run evaluation
%run ./scripts/arm_evaluation_main.py --cedar-api-key $cedar_api_key
Step 7: Analysis of results¶
The figures 9 and 10 in the paper were generated using an R script (generate_plots.R) that takes the .csv files with the results of the 8 previous experiments as the input and generates the following plots:
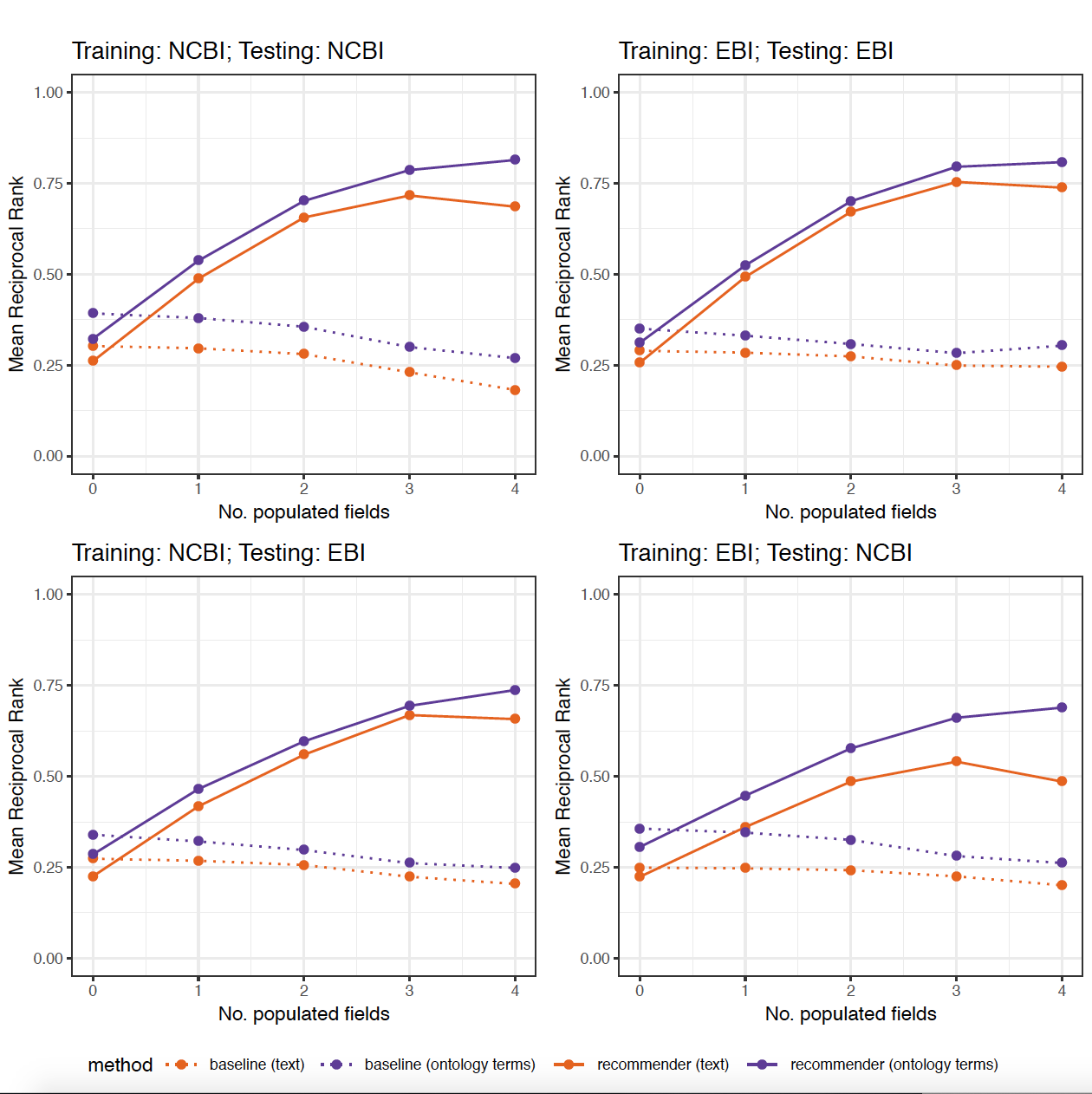
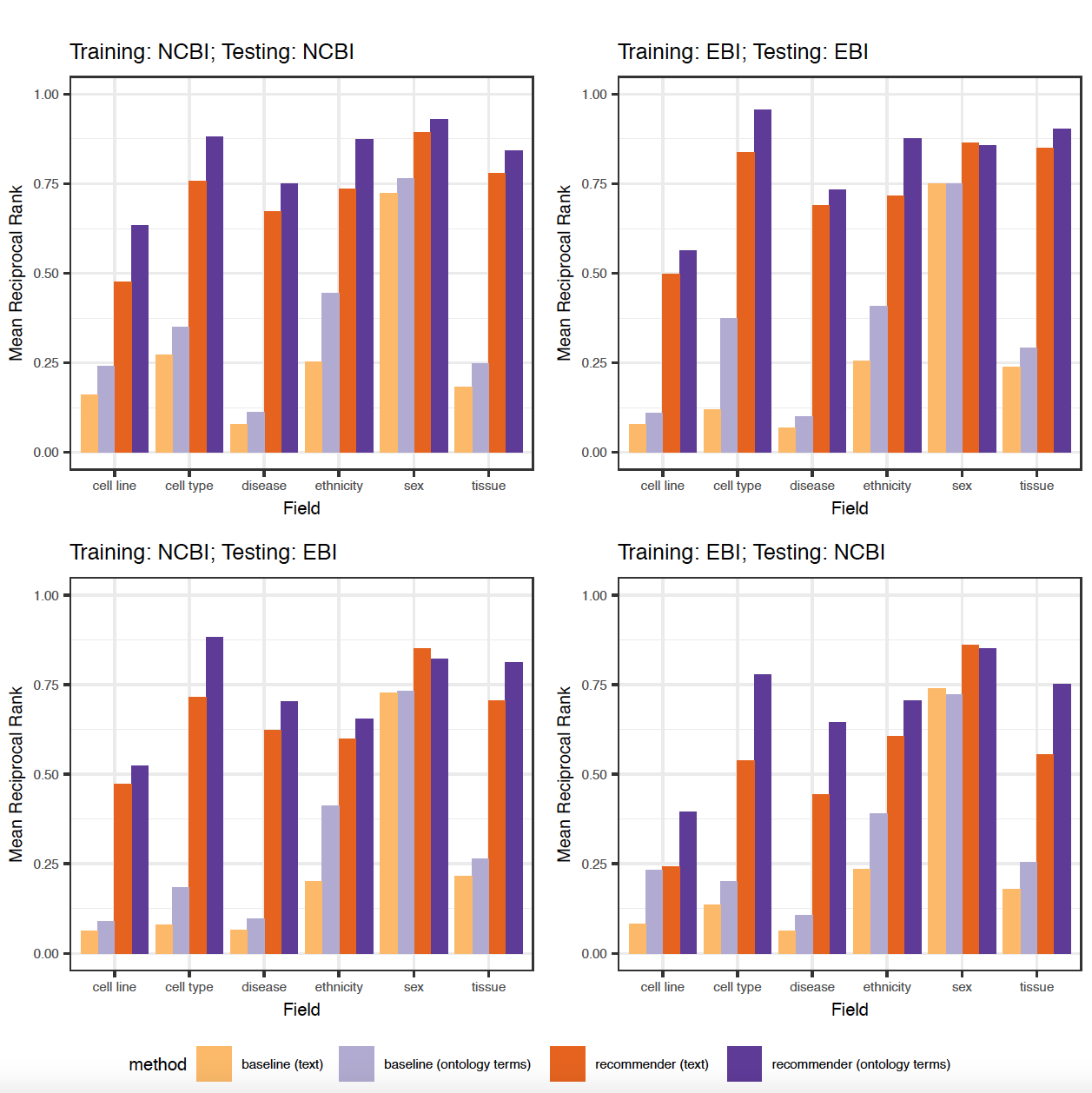
Additional experiments:¶
This section describes two additional experiments that we conducted to evaluate our approach.
Additional experiment #1¶
As described in the paper (Section 3.1.3), the values suggested for the target field are ranked according to a recommendation score that provides an absolute measurement of the goodness of the recommendation. The recommendation score is calculated according to the following expression:
$$recommendation\_score(v') = context\_matching\_score(r',C) * conf(r')$$where $v'$ is a value for the target field extracted from the consequent of a selected rule $r'$, $context\_matching\_score$ is the function that computes the context-matching score and $conf$ is the function that returns the confidence of a particular rule. Values with the same recommendation score are sorted by support before returning them to the user.
In this experiment, we wanted to evaluate the impact of replacing confidence by a different metric known as lift. The lift metric measures the interestingness of importance of a rule and it is widely used in association rule mining. That is, we wanted to evaluate our system when the recommendation score is calculated as:
$$recommendation\_score(v') = context\_matching\_score(r',C) * lift(r')$$We also wanted to study the impact of using lift instead of support as a secondary criteria to rank the results that have the same recommendation score.
We conducted a new experiment based on metadata from the NCBI BioSample database. We reused the rules previously generated based on textual metadata from the NCBI BioSample database and 1,000 instances as the test set. The results obtained, which are shown in the following table, confirm that confidence performs better than support in our case. They also show that the second criterion, used to sort the results that have the same recommendation score, influences the final results minimally.
| Approach | 1st criterion | 2nd criterion | MRR (top 5) |
|---|---|---|---|
| Current approach | confidence | support | 0.54 |
| Alternative 1 | confidence | lift | 0.53 |
| Alternative 2 | lift | confidence | 0.32 |
| Alternative 3 | lift | support | 0.32 |
Available materials:¶
- Rules used [download]
- CEDAR instances (testing) [download]
- Results files:
- File 1 (confidence, support) [download]
- File 2 (confidence, lift) [download]
- File 3 (lift, confidence) [download]
- File 4 (lift, support) [download]
Additional experiment #2¶
The evaluation described in the paper is focused on a subset of 6 fields commonly used to describe human samples. In this experiment, we wanted to study the impact of using more fields on the performance of the system. We executed the rule generation process for a set of 5,000 NCBI BioSample instances using (1) the 6 fields used in our main evaluation; and (2) all the fields in the BioSample template (26 fields). Then, we tested the performance of those rules using a set of 500 instances. The rules were generated with a minimum confidence of 0.3 and a minimum support of 0.002.
When using 6 fields, the system produced 572 rules in 5.1 seconds, and obtained an MRR of 0.318. For 26 fields, the system generated 30,559 rules (ncbi-text-rules-additional-experiment-2) in 40.8 seconds and generated suggestions with an MRR of 0.315. The results show that adding more fields considerable increased the number of rules generated and the time needed to generate the rules, but the difference in the accuracy of the suggestions was minimal. Even though the number of rules for 26 fields is 53 times higher than the number of rules for 6 fields, our approach was able to identify the rules that best matched the context entered by the user and ignored the noise produced by other rules in the system.
| No. fields | No. rules generated | No. rules after filtering | Rules generation time (seg) | Mean recommendation time (ms) | MRR (top 5) |
|---|---|---|---|---|---|
| 6 | 775 | 572 | 5.10 | 43.64 | 0.318 |
| 26 (all) | 233,363 | 30,559 | 40.81 | 44.79 | 0.315 |
Available materials:¶
- CEDAR instances [download]
- Rules generated [download]
- Results files:
- Using 6 fields [download]
- Using 26 fields [download]