RNA-Seq data corresponding to BS-Seq Oyster Sperm sample¶
Shortcuts
Data
RNA-seq data from both the sperm and pooled gill samples were provided by the core facility.
Originally BAM files were provided, later followed up with fastq files.
%20Discovery%20Environment.png)
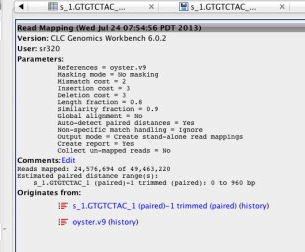
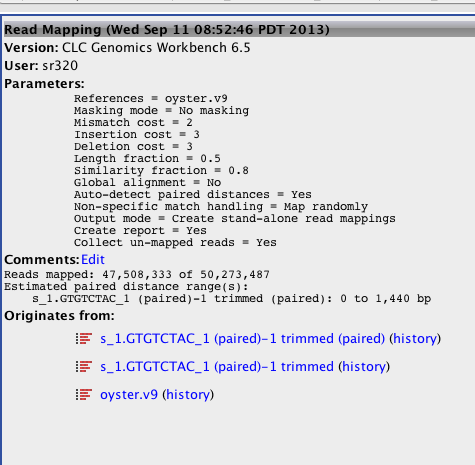
Sperm data = 72976/GTGTCTAC
CLC¶
fastq import (s_1.GTGTCTAC_1 (paired)-1)
51,084,360 sequences
Discard read names = Yes Discard quality scores = No Paired orientation = Paired reads (forward-reverse) Minimum distance = 180 Maximum distance = 250 Original sequence resource = /Volumes/NGS Drive/NGS Raw Data/RNA_complement_BS/C25GNACXX.oyster.fastqs/s_1.GTGTCTAC_1.fastq Original sequence resource = /Volumes/NGS Drive/NGS Raw Data/RNA_complement_BS/C25GNACXX.oyster.fastqs/s_1.GTGTCTAC_2.fastq Quality score = NCBI/Sanger or Illumina Pipeline 1.8 and later MiSeq de-multiplexing = No
QC Report¶
https://docs.google.com/file/d/0B9V_gF766XZAS0dRTE1LSDFBcXc/preview
Supp QC Report¶
https://docs.google.com/file/d/0B9V_gF766XZAZ2dYLTk3QmVvQVE/preview
Trimming

RNA-seq¶
Input file

Need to modify GFF so that CLC recognizes to annotate
http://eagle.fish.washington.edu/cnidarian/oyster.v9.gene_mRNA.gff
!head /Volumes/web/cnidarian/oyster.v9.gene_mRNA.gff
C16582 GLEAN gene 35 385 0.555898 - . ID=CGI_10000001; C16582 GLEAN mrna 35 385 . - 0 Parent=CGI_10000001; C17212 GLEAN gene 31 363 0.999572 + . ID=CGI_10000002; C17212 GLEAN mrna 31 363 . + 0 Parent=CGI_10000002; C17316 GLEAN gene 30 257 0.555898 + . ID=CGI_10000003; C17316 GLEAN mrna 30 257 . + 0 Parent=CGI_10000003; C17476 GLEAN gene 34 257 0.998947 - . ID=CGI_10000004; C17476 GLEAN mrna 104 257 . - 0 Parent=CGI_10000004; C17476 GLEAN mrna 34 74 . - 2 Parent=CGI_10000004; C17998 GLEAN gene 196 387 1 - . ID=CGI_10000005;
!wc /Volumes/web/cnidarian/oyster.v9.gene_mRNA.gff
224718 2022462 14376214 /Volumes/web/cnidarian/oyster.v9.gene_mRNA.gff
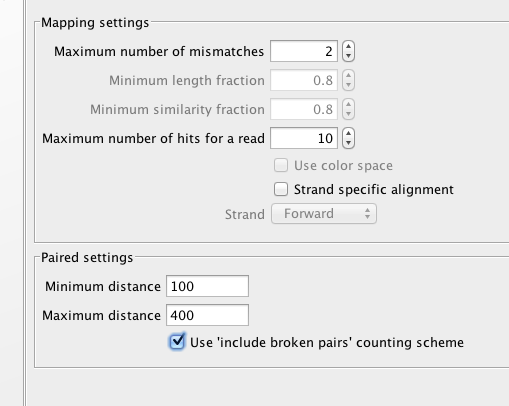
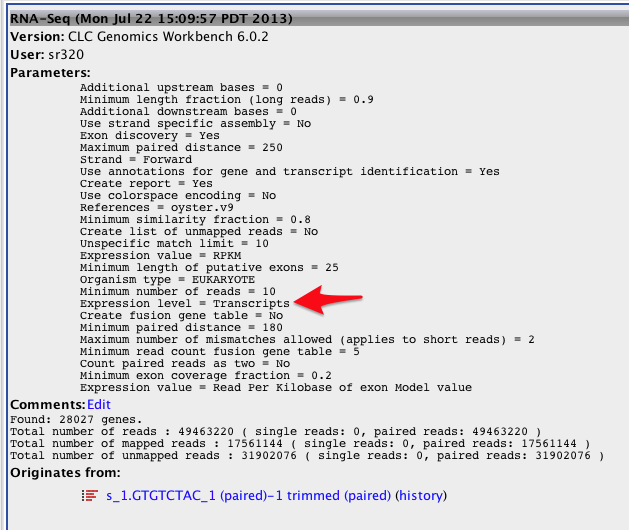
Annotated version 9 of oyster genome then ran RNA-seq with Exon Discovery and Expression value as transcripts. Only using paired data.

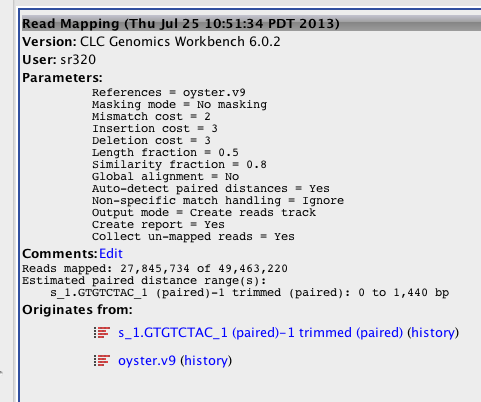
Concerned about the number of mapped reads¶
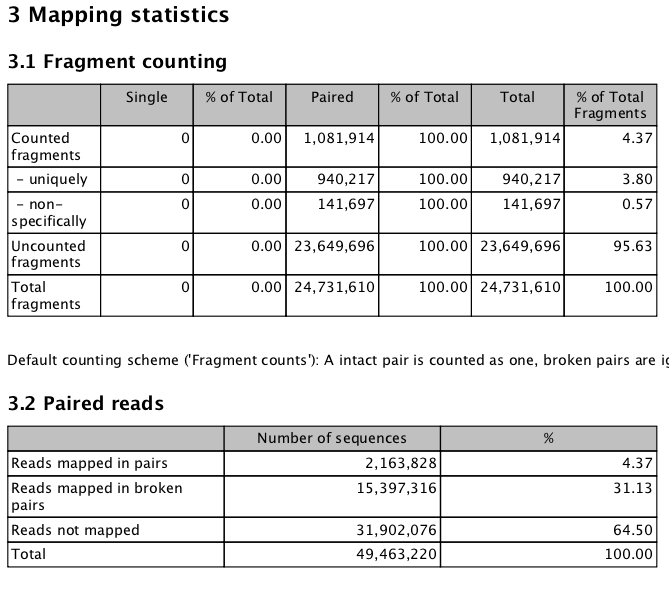
Full Report
http://eagle.fish.washington.edu/cnidarian/BiGo_RNAseq_report_072313a
Redux on RNA-seq on annotated genome¶
#September 12, 2013
#export of transcript-based table
!wc /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_1.txt
196692 2753710 15000644 /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_1.txt
!head /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_1.txt
"Feature ID" "Expression values" "Gene name" "Transcripts annotated" "Transcript length" "Transcript ID" "Unique transcript reads" "Total transcript reads" "Ratio of unique to total (transcript reads)" "Exons" "RPKM" "Relative RPKM" "Chromosome" "Chromosome region start" "Chromosome region end" CGI_10000780.1 0 CGI_10000780 2 1248 0 0 ? 1 0 1 scaffold350 1959 3206 CGI_10000780.2 0 CGI_10000780 2 102 0 0 ? 1 0 1 scaffold350 1105 1206 CGI_10000456.1 0 CGI_10000456 4 97 0 0 ? 1 0 1 scaffold28720 349 445 CGI_10000456.2 0 CGI_10000456 4 151 0 0 ? 1 0 1 scaffold28720 943 1093 CGI_10000456.3 0 CGI_10000456 4 124 0 0 ? 1 0 1 scaffold28720 1481 1604 CGI_10000456.4 0 CGI_10000456 4 66 0 0 ? 1 0 1 scaffold28720 1973 2038 CGI_10000457.1 0 CGI_10000457 7 94 0 0 ? 1 0 1 scaffold28720 5599 5692 CGI_10000457.2 0 CGI_10000457 7 138 0 0 ? 1 0 1 scaffold28720 5175 5312 CGI_10000457.3 0 CGI_10000457 7 50 0 0 ? 1 0 1 scaffold28720 4978 5027
from pandas import *
# read data from data file into a pandas DataFrame
BiGoRNAseq_exon = read_table("/Volumes/web/cnidarian/BiGoRNAseq_exon_exp_1.txt", # name of the data file
#sep=",", # what character separates each column?
na_values=["", " "]) # what values should be considered "blank" values?
--------------------------------------------------------------------------- IOError Traceback (most recent call last) <ipython-input-2-9169b38b7d32> in <module>() 4 BiGoRNAseq_exon = read_table("/Volumes/web/cnidarian/BiGoRNAseq_exon_exp_1.txt", # name of the data file 5 #sep=",", # what character separates each column? ----> 6 na_values=["", " "]) # what values should be considered "blank" values? /Users/sr320/anaconda/lib/python2.7/site-packages/pandas/io/parsers.pyc in parser_f(filepath_or_buffer, sep, dialect, compression, doublequote, escapechar, quotechar, quoting, skipinitialspace, lineterminator, header, index_col, names, prefix, skiprows, skipfooter, skip_footer, na_values, true_values, false_values, delimiter, converters, dtype, usecols, engine, delim_whitespace, as_recarray, na_filter, compact_ints, use_unsigned, low_memory, buffer_lines, warn_bad_lines, error_bad_lines, keep_default_na, thousands, comment, decimal, parse_dates, keep_date_col, dayfirst, date_parser, memory_map, nrows, iterator, chunksize, verbose, encoding, squeeze) 399 buffer_lines=buffer_lines) 400 --> 401 return _read(filepath_or_buffer, kwds) 402 403 parser_f.__name__ = name /Users/sr320/anaconda/lib/python2.7/site-packages/pandas/io/parsers.pyc in _read(filepath_or_buffer, kwds) 207 208 # Create the parser. --> 209 parser = TextFileReader(filepath_or_buffer, **kwds) 210 211 if nrows is not None: /Users/sr320/anaconda/lib/python2.7/site-packages/pandas/io/parsers.pyc in __init__(self, f, engine, **kwds) 507 self.options['has_index_names'] = kwds['has_index_names'] 508 --> 509 self._make_engine(self.engine) 510 511 def _get_options_with_defaults(self, engine): /Users/sr320/anaconda/lib/python2.7/site-packages/pandas/io/parsers.pyc in _make_engine(self, engine) 609 def _make_engine(self, engine='c'): 610 if engine == 'c': --> 611 self._engine = CParserWrapper(self.f, **self.options) 612 else: 613 if engine == 'python': /Users/sr320/anaconda/lib/python2.7/site-packages/pandas/io/parsers.pyc in __init__(self, src, **kwds) 891 # #2442 892 kwds['allow_leading_cols'] = self.index_col is not False --> 893 self._reader = _parser.TextReader(src, **kwds) 894 895 # XXX /Users/sr320/anaconda/lib/python2.7/site-packages/pandas/_parser.so in pandas._parser.TextReader.__cinit__ (pandas/src/parser.c:2771)() /Users/sr320/anaconda/lib/python2.7/site-packages/pandas/_parser.so in pandas._parser.TextReader._setup_parser_source (pandas/src/parser.c:4803)() IOError: File /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_1.txt does not exist
BiGoRNAseq_exon.dtypes
print BiGoRNAseq_exon
--------------------------------------------------------------------------- NameError Traceback (most recent call last) <ipython-input-3-e9cda2cd03df> in <module>() ----> 1 print BiGoRNAseq_exon NameError: name 'BiGoRNAseq_exon' is not defined
BiGoRNAseq_exon['RPKM'].hist(bins=100);
#Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function.
axis([0, 1000, 0, 1000000])
--------------------------------------------------------------------------- NameError Traceback (most recent call last) <ipython-input-4-b327e52f5762> in <module>() ----> 1 BiGoRNAseq_exon['RPKM'].hist(bins=100); 2 #Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function. 3 axis([0, 1000, 0, 1000000]) NameError: name 'BiGoRNAseq_exon' is not defined
BiGoRNAseq_exon(x='Transcripts annotated', y='Transcripts length', style='o');
--------------------------------------------------------------------------- TypeError Traceback (most recent call last) <ipython-input-44-35489418b778> in <module>() ----> 1 BiGoRNAseq_exon(x='Transcripts annotated', y='Transcripts length', style='o'); TypeError: 'DataFrame' object is not callable
Upload to SQLShare¶
Query to get Genome track
SELECT
["Chromosome"] as seqname,
'CLC_RNAseq' as source,
'TranscriptExp' as feature,
["Chromosome region start"] as start,
["Chromosome region start"] as [end],
["RPKM"] as score,
'.' as strand,
'.' as frame,
["Gene name"] as attribute
FROM [sr320@washington.edu].[table_BiGoRNAseq_exon_exp_1.txt]
!head /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track.csv
!tr ',' "\t" </Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track.csv> /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track.gff
!head /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track.gff
!wc /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track.gff
196692 1770228 14001279 /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track.gff
!head /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track2.csv
!tr ',' "\t" </Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track2.csv> /Volumes/web/cnidarian/BiGoRNAseq_exon_exp_track2.igv
IGV file based on exons / transcripts
http://eagle.fish.washington.edu/cnidarian/BiGoRNAseq_exon_exp_track2.sorted.igv
#WHY not just run bedtools on Accepted hit bam?
RNA-seq on Genes Only¶
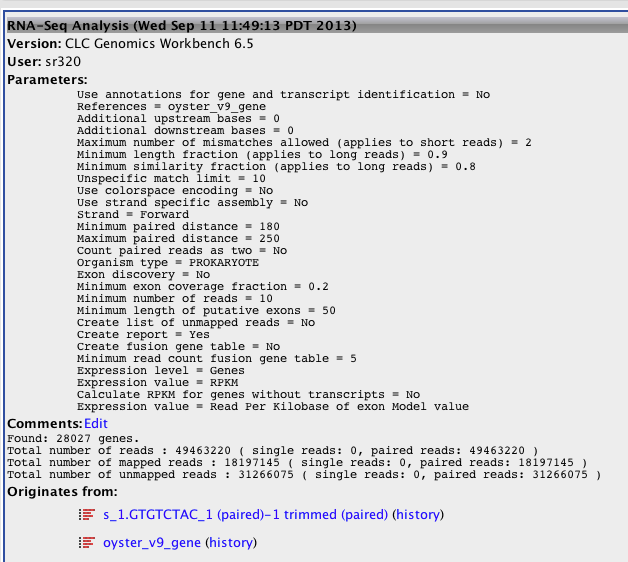
!head /Volumes/web/cnidarian/BiGo_RNAseq_genes
"Feature ID" "Expression values" "Gene length" "Unique gene reads" "Total gene reads" "RPKM" CGI_10000780 0 1350 0 0 0 CGI_10000456 7.892 438 8 9 7.892 CGI_10000457 7.643 603 6 12 7.643 CGI_10000774 0 375 0 0 0 CGI_10000917 0 426 0 0 0 CGI_10000861 0 2004 0 0 0 CGI_10000994 16.913 1635 64 72 16.913 CGI_10000643 0.696 552 1 1 0.696 CGI_10000763 0 249 0 0 0
RefMap (to see what proportion map)¶
Report
http://eagle.fish.washington.edu/cnidarian/BiGo_RNA_refmap_report_a
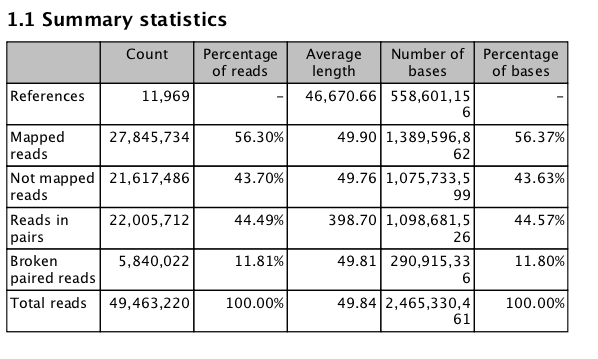
Only about 50% of reads map?
Will run again an try to assemble unmapped reads to determine what they are..
Running- reduced stringency to default, ignore vs random, kept unmapped reads, and output set as track
REDUX on Reference assembly¶
#September 11 2013
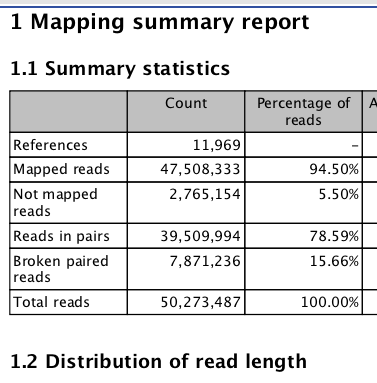
Big Difference! Seems to be the fact that mapped non-specific reads randomly....
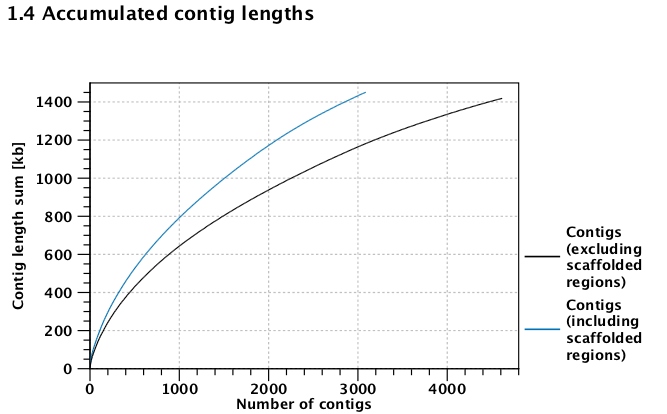
!tail /Volumes/web/cnidarian/BiGo_RNAseq_unmapped.fa
>s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_3093 GTGGTGCTCGGTCTGTATAACATCTGGACAGACTCCTGTTTTGTCAATCAGTTCTTGTAT GTGGTGTTGAAGAAAGTTAGAAACTCCAATATTCTTTACTTTACCTTCCTTCTGTAACTT GATCAGATCCCGCCAGCTCCCCACCCTCAGCTCCCCCTGATCTGGGTCATCTGGTTTCTT GCCCTGGGTCCCTGGCCAGTGAATCAGGTACAGGTCCAGGTACTGGCAGTCTAAGTTAG >s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_3094 GGGCTGCACAGATCTCTCAACAATCTCTTTCGTGAGAGTTAAGCGACTTTTAGGTTTGTT GTACATAACTGACAACAAACAGACAAAAGAACTTGGATTTTGGCACGGGGTTTCACTGTG TTACAAAGATCATGATACATTTTTGAGCAAATATTTTCACAACTGCATGAAAAATACATG TTTTTAACCGTTGGTGCCCTTGGTTGCATAGGTTGCCGCCATTTTGAAACATAGGAAGC
!blastx -query /Volumes/web/cnidarian/BiGo_RNAseq_unmapped.fa -db /Volumes/web/whale/fish546/blast/db/swissprot -out /Volumes/web/cnidarian/BiGoRNAseq_unmapped_swissprot_blastout -outfmt 6 -evalue 1E-10 -max_target_seqs 1 -num_threads 2
Selenocysteine (U) at position 690 replaced by X Selenocysteine (U) at position 690 replaced by X Selenocysteine (U) at position 690 replaced by X Selenocysteine (U) at position 140 replaced by X Selenocysteine (U) at position 196 replaced by X Selenocysteine (U) at position 204 replaced by X Selenocysteine (U) at position 196 replaced by X Selenocysteine (U) at position 193 replaced by X Selenocysteine (U) at position 493 replaced by X Selenocysteine (U) at position 24 replaced by X Selenocysteine (U) at position 63 replaced by X Selenocysteine (U) at position 25 replaced by X Selenocysteine (U) at position 63 replaced by X Selenocysteine (U) at position 60 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 73 replaced by X Selenocysteine (U) at position 43 replaced by X Selenocysteine (U) at position 73 replaced by X
!head /Volumes/web/cnidarian/BiGoRNAseq_unmapped_swissprot_blastout
s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_1 gi|3121895|sp|Q37705.1|COX1_ARTSF 55.23 507 224 2 4105 2594 5 511 2e-140 457 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_4 gi|3914777|sp|O61463.1|RLA2_CRYST 56.25 112 47 1 392 57 1 110 9e-21 86.7 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_5 gi|54041577|sp|P13418.2|POLS_CRPVC 22.81 754 440 25 2769 787 43 747 1e-25 118 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_6 gi|135489|sp|P11833.1|TBB_PARLI 99.29 420 3 0 1425 166 11 430 0.0 839 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_7 gi|341941223|sp|P29341.2|PABP1_MOUSE 85.23 88 13 0 1063 800 536 623 2e-38 150 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_8 gi|2500286|sp|Q26481.3|RL5_STYCL 75.93 295 70 1 1168 284 1 294 1e-134 395 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_9 gi|109940067|sp|P00428.2|COX5B_BOVIN 52.94 51 23 1 430 281 72 122 7e-13 65.9 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_10 gi|71151870|sp|Q8TC29.1|ENKUR_HUMAN 49.79 235 112 2 891 187 26 254 1e-67 218 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_13 gi|115305840|sp|Q2NKZ1.1|TCPH_BOVIN 82.76 116 20 0 1089 742 299 414 3e-108 212 s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_14 gi|6094094|sp|O57592.3|RL7A_FUGRU 77.08 240 53 1 770 51 29 266 1e-125 366
!wc /Volumes/web/cnidarian/BiGoRNAseq_unmapped_swissprot_blastout
1600 19200 271972 /Volumes/web/cnidarian/BiGoRNAseq_unmapped_swissprot_blastout
##Blast againa and get taxonomy info
# ./blastn -query /Volumes/web/cnidarian/BiGo_RNAseq_unmapped.fa -db /Volumes/CLC_blastdatabases/nt -out /Volumes/web/cnidarian/BiGo_RNAseq_unmapped_nt_blastout_taxa2 -outfmt "6 std stitle staxids sscinames scomnames sblastnames" -evalue 1E-20 -max_target_seqs 1 -task blastn -num_threads 6
^C
!head /Volumes/web/cnidarian/BiGo_RNAseq_unmapped_nt_blastout_taxa2
s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_1 gi|6636083|gb|AF177226.1|AF177226 99.54 11122 47 2 1476 12593 18224 7103 0.0 1.982e+04 Crassostrea gigas mitochondrial DNA, complete genome 29159 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_1 gi|6636083|gb|AF177226.1|AF177226 99.86 1474 2 0 2 1475 1474 1 0.0 2650 Crassostrea gigas mitochondrial DNA, complete genome 29159 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_2 gi|187762791|gb|EU672831.1| 99.56 901 4 0 4 904 8445 7545 0.0 1608 Crassostrea gigas isolate ORCg-4 mitochondrion, complete genome 29159 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_3 gi|375073746|gb|JF744732.1| 72.50 240 49 5 36 264 326 93 1e-26 129 Ostrea edulis clone M13-F.OEI1 CD650310-like protein mRNA, partial sequence 37623 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_4 gi|212374119|emb|FM165434.1| 74.27 342 82 2 54 392 339 1 5e-53 217 Mytilus galloprovincialis mRNA for ribosomal protein (p2 gene) 29158 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_6 gi|194068374|dbj|AB374929.1| 95.00 1260 63 0 166 1425 1356 97 0.0 1988 Saccostrea kegaki mRNA for beta-tubulin, complete cds 182713 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_6 gi|194068374|dbj|AB374929.1| 74.74 190 12 4 5 158 1613 1424 1e-29 141 Saccostrea kegaki mRNA for beta-tubulin, complete cds 182713 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_7 gi|224081794|ref|XM_002196958.1| 77.39 283 61 1 800 1079 1857 1575 1e-53 221 PREDICTED: Taeniopygia guttata similar to poly A binding protein, cytoplasmic 4 (LOC100228528), mRNA 59729 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_8 gi|40642989|emb|AJ563456.1| 99.76 829 2 0 367 1195 829 1 0.0 1487 Crassostrea gigas partial mRNA for ribosomal protein L5 (rpl5 gene) 29159 N/A N/A N/A s_1.GTGTCTAC_1_(paired)-1_trimmed_(paired)_un-mapped_reads_[no_read_group]_(single)_contig_10 gi|259013499|ref|NM_001165022.1| 69.58 503 147 3 224 723 863 364 2e-50 210 Saccoglossus kowalevskii enkurin, TRPC channel interacting protein (enkur), mRNA 10224 N/A N/A N/A
###Get Taxonomic Distribution
#SQLSshare
SELECT [Column14], count([Column1])
From
[sr320@washington.edu].[BiGo_RNAseq_unmapped_nt_blastout_taxa2]
Group by Column14
``
!head /Volumes/web/cnidarian/BiGoRNAseq_taxid_unmapped.csv
!wc /Volumes/web/cnidarian/BiGoRNAseq_taxid_unmapped.csv
142 142 1308 /Volumes/web/cnidarian/BiGoRNAseq_taxid_unmapped.csv
from pandas import *
# read data from data file into a pandas DataFrame
BiGoRNAsequm = read_csv("http://eagle.fish.washington.edu/cnidarian/BiGoRNAseq_taxid_unmapped.csv", # name of the data file
sep=",", # what character separates each column?
na_values=["", " "]) # what values should be considered "blank" values?
print BiGoRNAsequm
<class 'pandas.core.frame.DataFrame'> Int64Index: 141 entries, 0 to 140 Data columns: Column14 141 non-null values Unnamed: 1 141 non-null values dtypes: int64(2)
BiGoRNAsequm['Unnamed: 1'].hist(bins=50);
#Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function.
#plt.axis([0, 1, 0, 40])
<matplotlib.axes.AxesSubplot at 0x11eb9e090>

@fu
File "<ipython-input-10-93faf69d27a7>", line 1 @fu ^ SyntaxError: unexpected EOF while parsing
Tophat Analysis¶
--
%20Discovery%20Environment.png)
TopHat Output
The tophat script produces a number of files in the directory in which it was invoked. Most of these files are internal, intermediate files that are generated for use within the pipeline. The output files you will likely want to look at are:
accepted_hits.bam. A list of read alignments in SAM format. SAM is a compact short read alignment format that is increasingly being adopted. The formal specification is here.
junctions.bed. A UCSC BED track of junctions reported by TopHat. Each junction consists of two connected BED blocks, where each block is as long as the maximal overhang of any read spanning the junction. The score is the number of alignments spanning the junction.
insertions.bed and deletions.bed. UCSC BED tracks of insertions and deletions reported by TopHat.
Insertions.bed - chromLeft refers to the last genomic base before the insertion.
Deletions.bed - chromLeft refers to the first genomic base of the deletion.
Data @ http://eagle.fish.washington.edu/cnidarian/index.php?dir=tophat_071313%2F
ls /Volumes/web/cnidarian/tophat_071313
accepted_hits.bam* prep_reads.info* deletions.bed* s_1.bam* insertions.bed* s_1.bam.bai* junctions.bed* unmapped.bam* lookup_roberts_grc_oyster.xls*
!head /Volumes/web/cnidarian/tophat_071313/junctions.bed
track name=junctions description="TopHat junctions" C14162 6 239 JUNC00000001 12 - 6 239 255,0,0 2 36,49 0,184 C14832 78 229 JUNC00000002 4 - 78 229 255,0,0 2 43,27 0,124 C17940 206 349 JUNC00000003 1 - 206 349 255,0,0 2 31,19 0,124 C17998 32 262 JUNC00000004 734 - 32 262 255,0,0 2 49,49 0,181 C18186 378 530 JUNC00000005 1 + 378 530 255,0,0 2 29,21 0,131 C18450 359 554 JUNC00000006 6 + 359 554 255,0,0 2 38,49 0,146 C18694 57 245 JUNC00000007 1 - 57 245 255,0,0 2 30,20 0,168 C18754 51 472 JUNC00000008 6 + 51 472 255,0,0 2 47,32 0,389 C19028 89 279 JUNC00000009 76 + 89 279 255,0,0 2 40,49 0,141

URL to load IGV Session corresponding to screenshot above.
http://eagle.fish.washington.edu/cnidarian/BiGo_RNAseq_072213_igv_session.xml
Using Samtools to interogate TopHat Bam file¶
reference: http://left.subtree.org/2012/04/13/counting-the-number-of-reads-in-a-bam-file/
cd /Volumes/Bay3/Software/BSMAP/bsmap-2.74/samtools
/Volumes/Bay3/Software/BSMAP/bsmap-2.74/samtools
ls
AUTHORS bam_index.o bedidx.o kstring.c COPYING bam_lpileup.c bgzf.c kstring.h ChangeLog bam_lpileup.o bgzf.h kstring.o INSTALL bam_mate.c bgzf.o libbam.a Makefile bam_mate.o bgzip.c misc/ Makefile.mingw bam_md.c cut_target.c phase.c NEWS bam_md.o cut_target.o phase.o bam.c bam_pileup.c errmod.c razf.c bam.h bam_pileup.o errmod.h razf.h bam.o bam_plcmd.c errmod.o razf.o bam2bcf.c bam_plcmd.o examples/ razip.c bam2bcf.h bam_reheader.c faidx.c sam.c bam2bcf.o bam_reheader.o faidx.h sam.h bam2bcf_indel.c bam_rmdup.c faidx.o sam.o bam2bcf_indel.o bam_rmdup.o kaln.c sam_header.c bam2depth.c bam_rmdupse.c kaln.h sam_header.h bam2depth.o bam_rmdupse.o kaln.o sam_header.o bam_aux.c bam_sort.c khash.h sam_view.c bam_aux.o bam_sort.o klist.h sam_view.o bam_cat.c bam_stat.c knetfile.c sample.c bam_cat.o bam_stat.o knetfile.h sample.h bam_color.c bam_tview.c knetfile.o sample.o bam_color.o bam_tview.o kprobaln.c samtools* bam_endian.h bamtk.c kprobaln.h samtools.1 bam_import.c bamtk.o kprobaln.o win32/ bam_import.o bcftools/ kseq.h bam_index.c bedidx.c ksort.h
!samtools
/bin/sh: samtools: command not found
cp samtools /usr/local/bin
!samtools
Program: samtools (Tools for alignments in the SAM format)
Version: 0.1.18 (r982:295)
Usage: samtools <command> [options]
Command: view SAM<->BAM conversion
sort sort alignment file
mpileup multi-way pileup
depth compute the depth
faidx index/extract FASTA
index index alignment
idxstats BAM index stats (r595 or later)
fixmate fix mate information
flagstat simple stats
calmd recalculate MD/NM tags and '=' bases
merge merge sorted alignments
rmdup remove PCR duplicates
reheader replace BAM header
cat concatenate BAMs
targetcut cut fosmid regions (for fosmid pool only)
phase phase heterozygotes
!samtools view -c /Volumes/web/cnidarian/tophat_071313/s_1.bam
164718919
#only mapped reads
!samtools view -c -F 4 /Volumes/web/cnidarian/tophat_071313/s_1.bam
164718919
#unmapped reads
!samtools view -c -f 4 /Volumes/web/cnidarian/tophat_071313/s_1.bam
0
!samtools view -c /Volumes/web/cnidarian/tophat_071313/accepted_hits.bam
164718919
!samtools view -c /Volumes/web/cnidarian/tophat_071313/unmapped.bam
7263587
!samtools flagstat /Volumes/web/cnidarian/tophat_071313/s_1.bam
164718919 + 0 in total (QC-passed reads + QC-failed reads) 0 + 0 duplicates 164718919 + 0 mapped (100.00%:nan%) 164718919 + 0 paired in sequencing 82181831 + 0 read1 82537088 + 0 read2 42418114 + 0 properly paired (25.75%:nan%) 161646608 + 0 with itself and mate mapped 3072311 + 0 singletons (1.87%:nan%) 1264082 + 0 with mate mapped to a different chr 395312 + 0 with mate mapped to a different chr (mapQ>=5) 164718919 + 0 in total (QC-passed reads + QC-failed reads) 0 + 0 duplicates 164718919 + 0 mapped (100.00%:nan%) 164718919 + 0 paired in sequencing 82181831 + 0 read1 82537088 + 0 read2 42418114 + 0 properly paired (25.75%:nan%) 161646608 + 0 with itself and mate mapped 3072311 + 0 singletons (1.87%:nan%) 1264082 + 0 with mate mapped to a different chr 395312 + 0 with mate mapped to a different chr (mapQ>=5)
!samtools flagstat /Volumes/web/cnidarian/tophat_071313/accepted_hits.bam
164718919 + 0 in total (QC-passed reads + QC-failed reads) 0 + 0 duplicates 164718919 + 0 mapped (100.00%:nan%) 164718919 + 0 paired in sequencing 82181831 + 0 read1 82537088 + 0 read2 42418114 + 0 properly paired (25.75%:nan%) 161646608 + 0 with itself and mate mapped 3072311 + 0 singletons (1.87%:nan%) 1264082 + 0 with mate mapped to a different chr 395312 + 0 with mate mapped to a different chr (mapQ>=5)
!samtools flagstat /Volumes/web/cnidarian/tophat_071313/unmapped.bam
7161175 + 102412 in total (QC-passed reads + QC-failed reads) 0 + 0 duplicates 0 + 0 mapped (0.00%:0.00%) 7161175 + 102412 paired in sequencing 3670234 + 76916 read1 3490941 + 25496 read2 0 + 0 properly paired (0.00%:0.00%) 0 + 0 with itself and mate mapped 0 + 0 singletons (0.00%:0.00%) 0 + 0 with mate mapped to a different chr 0 + 0 with mate mapped to a different chr (mapQ>=5)
Tophat Data Into Cufflinks (IPlant)¶
#cufflinks failed
Quality Trimming in iPlant¶
Pre-Process Complete, generating one file
Downloading and running through Fastx quality Trimmer
!wc /Volumes/web/cnidarian/BiGo_RNAseq_fastx_qual.fastq
190998292 190998292 6469564027 /Volumes/web/cnidarian/BiGo_RNAseq_fastx_qual.fastq
!wc /Volumes/web/cnidarian/BiGo_RNAseq_PreProcess.fastq
191927856 191927856 6501051882 /Volumes/web/cnidarian/BiGo_RNAseq_PreProcess.fastq 191927856 191927856 6501051882 /Volumes/web/cnidarian/BiGo_RNAseq_PreProcess.fastq
FastQC in iPlant¶
cd /Volumes/web/cnidarian/
/Volumes/web/cnidarian
!iget -r /iplant/home/sr320/analyses/FastQC_0.10.1__BiGoRNA-2014-03-24-10-33-44.916
Run on filtered file..
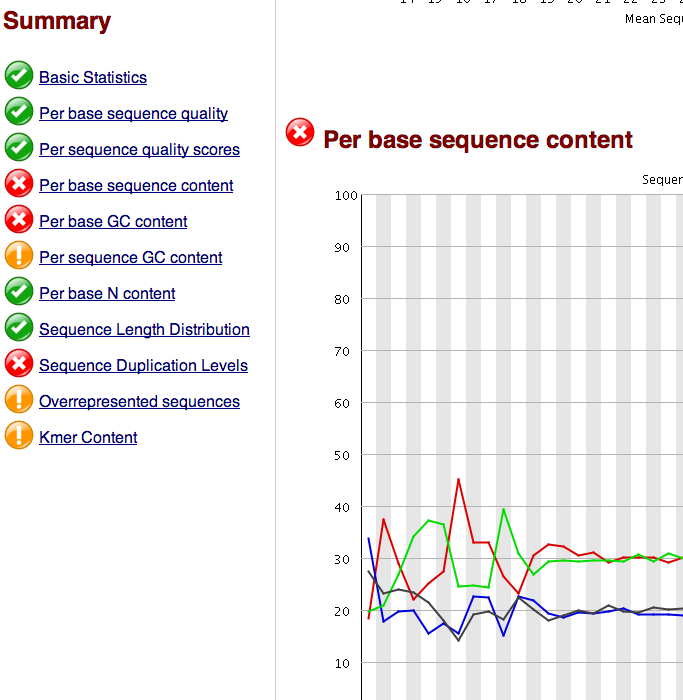
In CLC¶
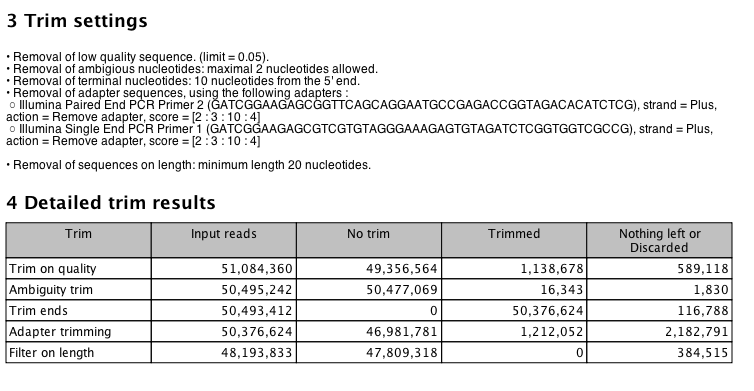
new file
!head /Volumes/web/cnidarian/BiGoRNA_genetable_clc
"Name" "Chromosome" "Region" "Expression value" "Gene length" "RPKM" "Unique gene reads" "Total gene reads" "Transcripts annotated" "Detected transcripts" "Exon length" "Exons" "Unique exon reads" "Total exon reads" "Ratio of unique to total (exon reads)" "Unique exon-exon reads" "Total exon-exon reads" "Unique intron reads" "Total intron reads" "Ratio of intron to total gene reads" CGI_10000780 scaffold350 complement(1105..3206) 0 2102 0 0 0 2 0 1350 2 0 0 ? 0 0 0 0 ? CGI_10000456 scaffold28720 349..2038 0 1690 0.323 2 2 4 0 438 4 0 0 ? 0 0 2 2 1 CGI_10000457 scaffold28720 complement(3692..5692) 1 2001 0.469 0 4 7 0 603 7 0 1 0 0 0 0 3 0.75 CGI_10000774 scaffold31684 complement(6549..7563) 0 1015 0.189 1 1 2 0 375 2 0 0 ? 0 0 1 1 1 CGI_10000917 scaffold32586 1736..2161 0 426 0 0 0 1 0 426 1 0 0 ? 0 0 0 0 ? CGI_10000861 scaffold900 complement(6516..8519) 1 2004 0.035 1 1 1 1 2004 1 1 1 1 0 0 0 0 0 CGI_10000994 scaffold33132 complement(5089..12821) 16 7733 1.385 27 32 11 2 1635 11 14 16 0.875 0 0 13 16 0.5 CGI_10000643 scaffold740 109..2125 0 2017 0 0 0 4 0 552 4 0 0 ? 0 0 0 0 ? CGI_10000763 scaffold31534 complement(1194..3724) 0 2531 0 0 0 3 0 249 3 0 0 ? 0 0 0 0 ?
#Location of SQLShare python tools: you can empty ("") if tools are in PATH
spd="/Users/sr320/sqlshare-pythonclient/tools/"
!python {spd}singleupload.py -d BiGoRNA_genetable_clc /Volumes/web/cnidarian/BiGoRNA_genetable_clc
processing chunk line 0 to 28028 (0.0644998550415 s elapsed) pushing /Volumes/web/cnidarian/BiGoRNA_genetable_clc... parsing FA491187... finished BiGoRNA_genetable_clc
%pylab inline
Populating the interactive namespace from numpy and matplotlib
import numpy as np
import matplotlib.pyplot as plt
from pandas import *
# read data from data file into a pandas DataFrame
BiGoRNA = read_table("/Volumes/web/cnidarian/BiGoRNA_genetable_clc", # name of the data file
#sep="\t", # what character separates each column?
#na_values=["", " "], # what values should be considered "blank" values?
#header=None
)
--------------------------------------------------------------------------- ImportError Traceback (most recent call last) <ipython-input-3-e36ec69cdc5b> in <module>() ----> 1 from pandas import * 2 3 # read data from data file into a pandas DataFrame 4 BiGoRNA = read_table("/Volumes/web/cnidarian/BiGoRNA_genetable_clc", # name of the data file 5 #sep="\t", # what character separates each column? ImportError: No module named pandas
BiGoRNA
plot.BiGoRNA["Expression value"]
#Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function.
#plt.axis([0, 200, 0, 30000]);
#plt.title('Expression value');
--------------------------------------------------------------------------- AttributeError Traceback (most recent call last) <ipython-input-58-6b8ded5f438e> in <module>() ----> 1 plot.BiGoRNA["Expression value"] 2 #Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function. 3 #plt.axis([0, 200, 0, 30000]); 4 #plt.title('Expression value'); AttributeError: 'function' object has no attribute 'BiGoRNA'
plt.hist(BiGoRNA["Total intron reads"], 20)
plt.axis([0, 100, 0, 2000])
plt.show()

#generate some data
x = BiGoRNA["Expression value"]
y = BiGoRNA["Total intron reads"]
#plot the data
plt.plot(x, y, 'bo')
plt.show()

fig = plt.loglog(x, y, 'rs')

fig = plt.semilogx(x, y, 'g^')

Annotation of genetable_clc in SQLShare¶