Analysis of RNA-Seq data that corresponds to oyster gill BS-Seq data¶
Number of Reads
Grooming - assuming 1.8 thus Sanger Doing this because Galaxy will not recognize
#Tophat complete, had to go back and quality trim individual files
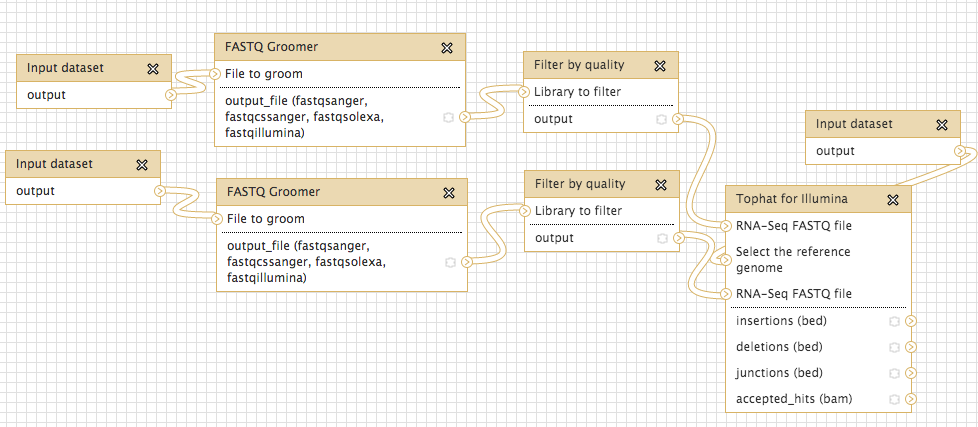
#checking accepted hits
!samtools view -c /Volumes/web/cnidarian/BiGill_RNAseqGalaxy_Tophat.bam
24876898
#only mapped reads
!samtools view -c -F 4 /Volumes/web/cnidarian/BiGill_RNAseqGalaxy_Tophat.bam
24876898
!samtools flagstat /Volumes/web/cnidarian/BiGill_RNAseqGalaxy_Tophat.bam
24876898 + 0 in total (QC-passed reads + QC-failed reads) 0 + 0 duplicates 24876898 + 0 mapped (100.00%:nan%) 24876898 + 0 paired in sequencing 12557606 + 0 read1 12319292 + 0 read2 108356 + 0 properly paired (0.44%:nan%) 1412054 + 0 with itself and mate mapped 23464844 + 0 singletons (94.32%:nan%) 0 + 0 with mate mapped to a different chr 0 + 0 with mate mapped to a different chr (mapQ>=5)
#Cufflinks needs GTF
!head /Volumes/web/cnidarian/TJGR_CDSgffread_filtered.gtf
# /usr/local2/cufflinks-1.0.3.Linux_x86_64/gffread oyster.v9.glean.final.rename.CDS.gff -o gffread_filtered.gtf ##gff-version 3 C16582 GLEAN mRNA 35 385 . - . ID=CGI_10000001 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001 C17212 GLEAN mRNA 31 363 . + . ID=CGI_10000002 C17212 GLEAN CDS 31 363 . + 0 Parent=CGI_10000002 C17316 GLEAN mRNA 30 257 . + . ID=CGI_10000003 C17316 GLEAN CDS 30 257 . + 0 Parent=CGI_10000003 C17476 GLEAN mRNA 34 257 . - . ID=CGI_10000004 C17476 GLEAN CDS 34 74 . - 2 Parent=CGI_10000004
Running cufflinks 1
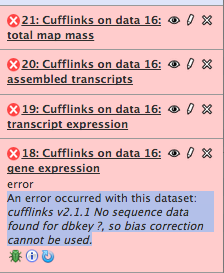
#hung for days; restart
Cufflinks worked in Galaxy¶
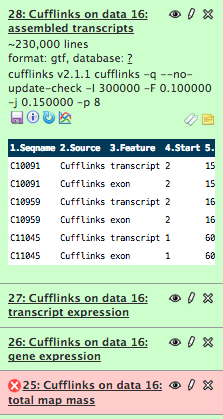
!head /Volumes/web/cnidarian/BiGillCufflinks_assembled_transcripts1.gtf
C10091 Cufflinks transcript 2 155 1000 . . gene_id "CUFF.1"; transcript_id "CUFF.1.1"; FPKM "212.5240682926"; frac "1.000000"; conf_lo "10.704568"; conf_hi "24.291136"; cov "167.595205"; C10091 Cufflinks exon 2 155 1000 . . gene_id "CUFF.1"; transcript_id "CUFF.1.1"; exon_number "1"; FPKM "212.5240682926"; frac "1.000000"; conf_lo "10.704568"; conf_hi "24.291136"; cov "167.595205"; C10959 Cufflinks transcript 2 162 1000 . . gene_id "CUFF.2"; transcript_id "CUFF.2.1"; FPKM "49.8667629708"; frac "1.000000"; conf_lo "1.969068"; conf_hi "9.451525"; cov "39.324630"; C10959 Cufflinks exon 2 162 1000 . . gene_id "CUFF.2"; transcript_id "CUFF.2.1"; exon_number "1"; FPKM "49.8667629708"; frac "1.000000"; conf_lo "1.969068"; conf_hi "9.451525"; cov "39.324630"; C11045 Cufflinks transcript 1 60 1000 . . gene_id "CUFF.3"; transcript_id "CUFF.3.1"; FPKM "28466.3975885016"; frac "1.000000"; conf_lo "29.588525"; conf_hi "66.574181"; cov "22448.430340"; C11045 Cufflinks exon 1 60 1000 . . gene_id "CUFF.3"; transcript_id "CUFF.3.1"; exon_number "1"; FPKM "28466.3975885016"; frac "1.000000"; conf_lo "29.588525"; conf_hi "66.574181"; cov "22448.430340"; C11484 Cufflinks transcript 3 116 1000 . . gene_id "CUFF.4"; transcript_id "CUFF.4.1"; FPKM "245.8763696681"; frac "1.000000"; conf_lo "3.893227"; conf_hi "16.129083"; cov "193.896630"; C11484 Cufflinks exon 3 116 1000 . . gene_id "CUFF.4"; transcript_id "CUFF.4.1"; exon_number "1"; FPKM "245.8763696681"; frac "1.000000"; conf_lo "3.893227"; conf_hi "16.129083"; cov "193.896630"; C11558 Cufflinks transcript 1 130 1000 . . gene_id "CUFF.5"; transcript_id "CUFF.5.1"; FPKM "332.4008375524"; frac "1.000000"; conf_lo "10.242182"; conf_hi "24.386147"; cov "262.129306"; C11558 Cufflinks exon 1 130 1000 . . gene_id "CUFF.5"; transcript_id "CUFF.5.1"; exon_number "1"; FPKM "332.4008375524"; frac "1.000000"; conf_lo "10.242182"; conf_hi "24.386147"; cov "262.129306";
This is a screenshot of file #28. interesting
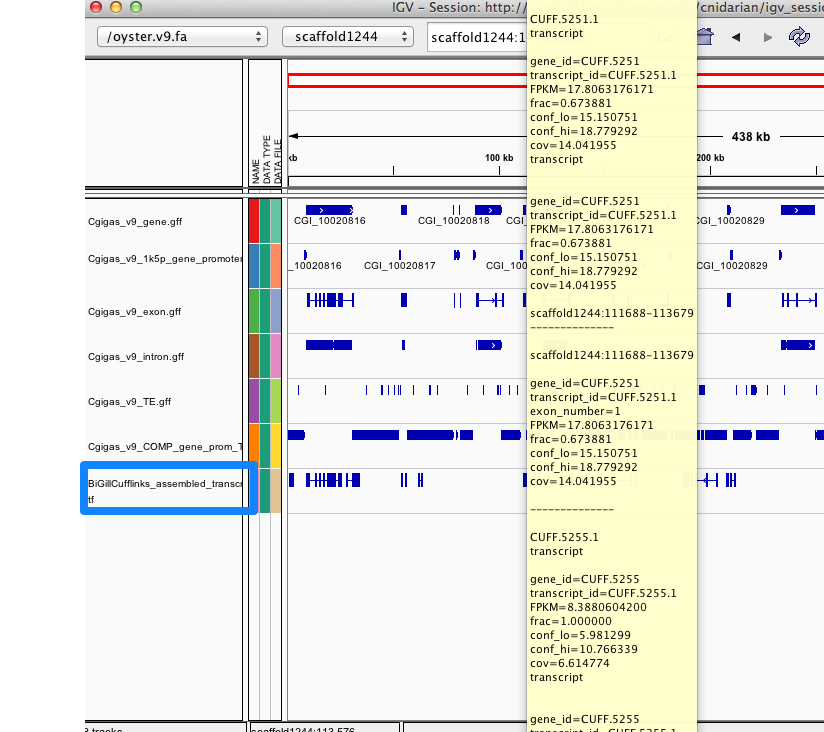
The two other files that cufflinks produced
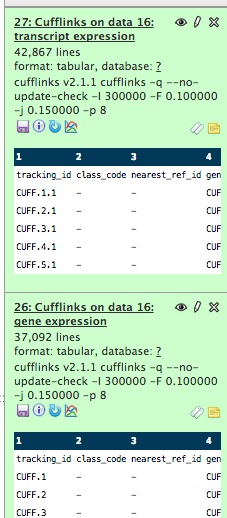
http://eagle.fish.washington.edu/cnidarian/BiGillCufflinks_transcripts_exp1.tabular
http://eagle.fish.washington.edu/cnidarian/BiGillCufflinks_gene_exp1.tabular
!wc /Volumes/web/cnidarian/BiGillCufflinks_transcripts_exp1.tabular
42867 557271 4118574 /Volumes/web/cnidarian/BiGillCufflinks_transcripts_exp1.tabular
!wc /Volumes/web/cnidarian/BiGillCufflinks_gene_exp1.tabular
37092 482196 3179694 /Volumes/web/cnidarian/BiGillCufflinks_gene_exp1.tabular
Using Bedtools to get exon coverage¶
!head /Volumes/web/cnidarian/BiGill_ThBAM_cov_exon_1.txt
C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 0 49 351 0.1396011 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 1 1 351 0.0028490 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 2 2 351 0.0056980 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 3 4 351 0.0113960 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 5 2 351 0.0056980 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 6 2 351 0.0056980 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 7 2 351 0.0056980 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 8 1 351 0.0028490 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 9 2 351 0.0056980 C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 10 2 351 0.0056980
from pandas import *
# read data from data file into a pandas DataFrame
ex = read_table("http://eagle.fish.washington.edu/cnidarian/BiGill_ThBAM_cov_exon_1.txt", # name of the data file
#sep=",", # what character separates each column?
na_values=["", " "]) # what values should be considered "blank" values?
print ex
#need to figure out column titles @fu
<class 'pandas.core.frame.DataFrame'> Int64Index: 2030332 entries, 0 to 2030331 Data columns: C16582 2030332 non-null values GLEAN 2030332 non-null values CDS 2030332 non-null values 35 2030332 non-null values 385 2030332 non-null values . 2020281 non-null values - 2020281 non-null values 0 2020281 non-null values Parent=CGI_10000001; 2020281 non-null values 0.1 2020281 non-null values 49 2020281 non-null values 351 2020281 non-null values 0.1396011 2020281 non-null values dtypes: float64(6), int64(1), object(6)
ex ['0.1396011'].hist(bins=50);
#Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function.
#plt.axis([0, 1, 0, 400000])
<matplotlib.axes.AxesSubplot at 0x107198e50>

!coveragebed -abam /Volumes/web/cnidarian/BiGill_RNAseqGalaxy_Tophat.bam -b /Volumes/web/trilobite/Crassostrea_gigas_v9_tracks/Cgigas_v9_exon.gff > /Volumes/web/cnidarian/BiGill_ThBAM_cov_exon_2.txt
!head /Volumes/web/cnidarian/BiGill_ThBAM_cov_exon_2.txt
C16582 GLEAN CDS 35 385 . - 0 Parent=CGI_10000001; 410 302 351 0.8603989 C17212 GLEAN CDS 31 363 . + 0 Parent=CGI_10000002; 16 314 333 0.9429429 C17316 GLEAN CDS 30 257 . + 0 Parent=CGI_10000003; 15 218 228 0.9561403 C17476 GLEAN CDS 104 257 . - 0 Parent=CGI_10000004; 2 73 154 0.4740260 C17476 GLEAN CDS 34 74 . - 2 Parent=CGI_10000004; 0 0 41 0.0000000 C17998 GLEAN CDS 196 387 . - 0 Parent=CGI_10000005; 0 0 192 0.0000000 C18346 GLEAN CDS 174 551 . + 0 Parent=CGI_10000009; 3577 378 378 1.0000000 C18428 GLEAN CDS 286 546 . - 0 Parent=CGI_10000010; 3474 261 261 1.0000000 C18964 GLEAN CDS 203 658 . - 0 Parent=CGI_10000011; 22 274 456 0.6008772 C18980 GLEAN CDS 30 674 . + 0 Parent=CGI_10000012; 15 446 645 0.6914729
#into SQLShare
SELECT Column1 as seqid,
Column4 as start,
Column5 as [end],
'BiGillExonExp' as Feature,
((cast([Column10]as float)/(cast([Column12]as float)))) as feat_bp
FROM [sr320@washington.edu].[BiGill_ThBAM_cov_exon_2.txt]
!head /Volumes/web/cnidarian/BiGill_ThBam_exon_feature.csv
!tr ',' "\t" </Volumes/web/cnidarian/BiGill_ThBam_exon_feature.csv> /Volumes/web/cnidarian/BiGill_ThBam_exon_feature.igv
!head /Volumes/web/cnidarian/BiGill_ThBam_exon_feature.igv
!head /Volumes/web/cnidarian/BiGill_ThBam_exon_feature.sorted.igv
seqid start end Feature feat_bp C16582 35 385 BiGillExonExp 1.16809116809117 C17212 31 363 BiGillExonExp 0.048048048048048 C17316 30 257 BiGillExonExp 0.0657894736842105 C17476 34 74 BiGillExonExp 0 C17476 104 257 BiGillExonExp 0.012987012987013 C17998 196 387 BiGillExonExp 0 C18346 174 551 BiGillExonExp 9.46296296296296 C18428 286 546 BiGillExonExp 13.3103448275862 C18964 203 658 BiGillExonExp 0.0482456140350877
SELECT
Gene,
count(feat_bp) as count,
stdev(feat_bp) as sd,
sum(feat_bp) as sum,
(stdev(feat_bp))/(sum(feat_bp)) as cv
FROM [sr320@washington.edu].[BiGill_RNAseq_exon]
Group by Gene
Having sum(feat_bp) > 0
!head /Volumes/web/cnidarian/BiGill_exonexp_gene.csv
!tr ',' "\t" </Volumes/web/cnidarian/BiGill_exonexp_gene.csv> /Volumes/web/cnidarian/BiGill_exonexp_gene.txt
!head /Volumes/web/cnidarian/BiGill_exonexp_gene.txt
sort: invalid number at field start: invalid count at start of `POS5'
#need to make igv file
#clean up Gene name and join
#find tails of genes with uniform versus variable expression @fu
Make Basic BiGill methylated CG (>50%) feature track¶
!head /Volumes/web/cnidarian/BiGill_mCG_feature.csv
!tr ',' "\t" </Volumes/web/cnidarian/BiGill_mCG_feature.csv> /Volumes/web/cnidarian/BiGill_mCG_feature.igv
!samtools sort -h
sort: illegal option -- h Usage: samtools sort [-on] [-m <maxMem>] <in.bam> <out.prefix>
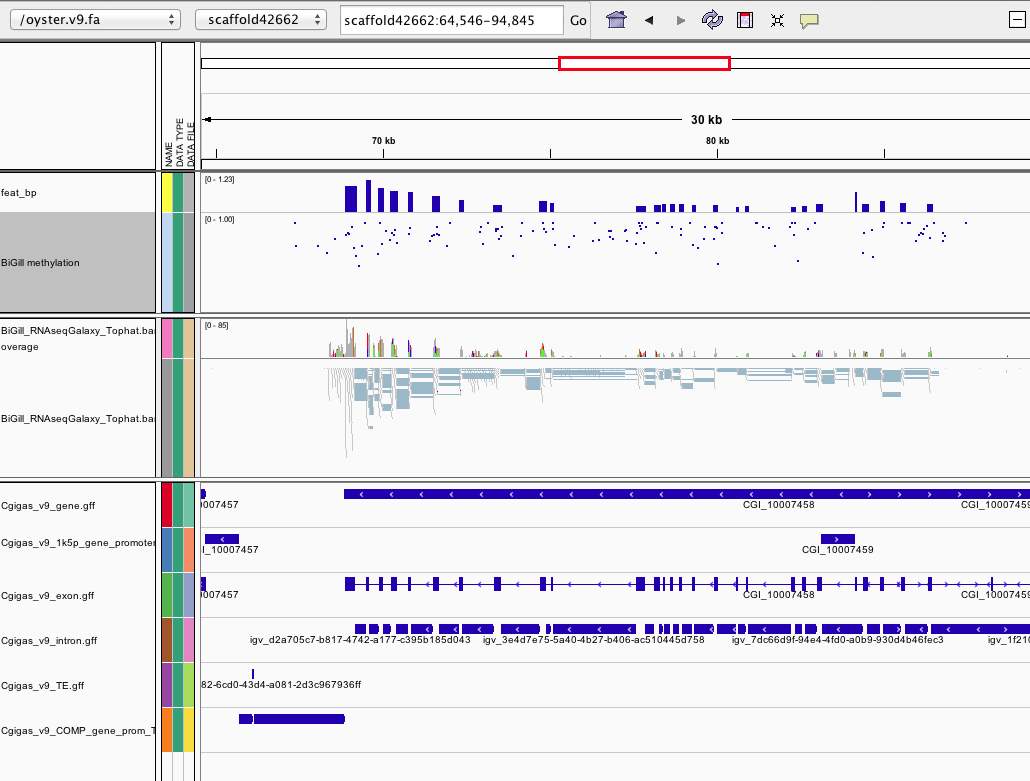
link
http://eagle.fish.washington.edu/cnidarian/igv_session_080513.xml
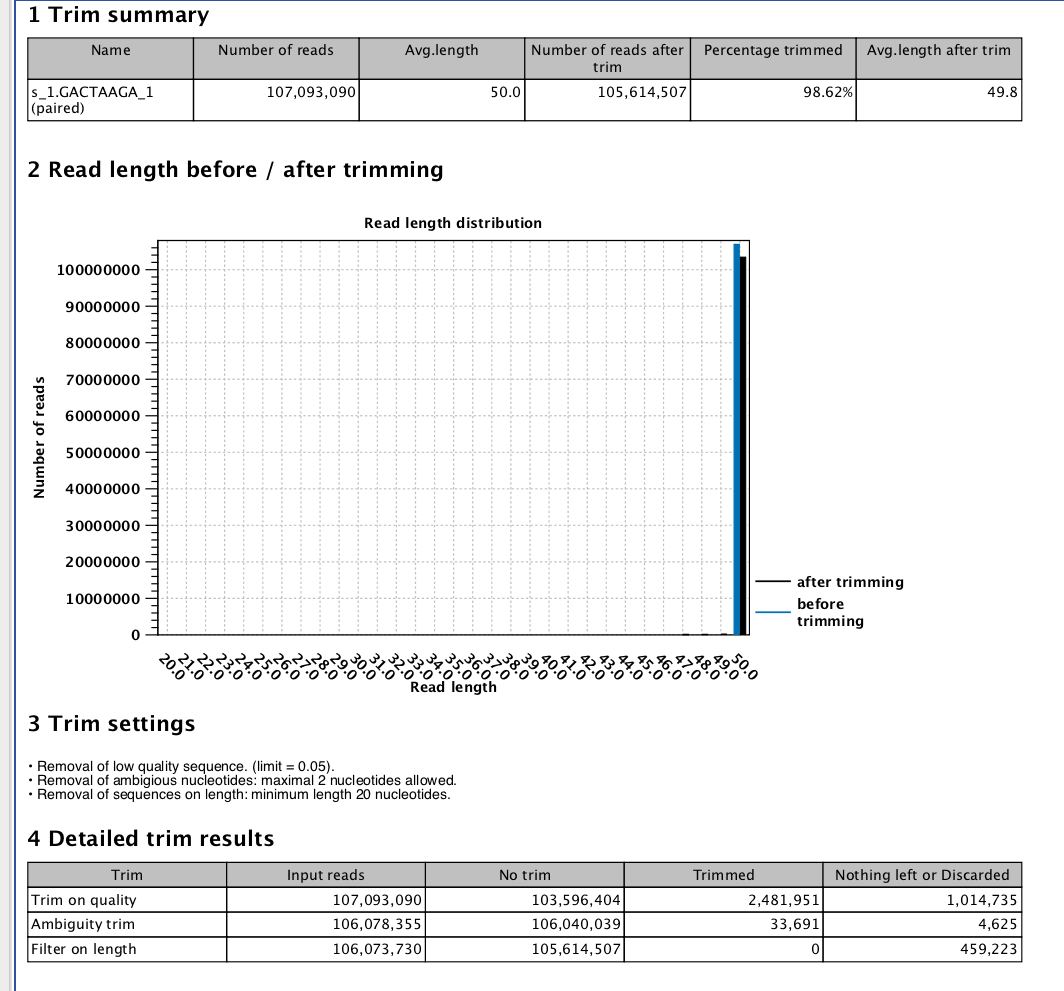
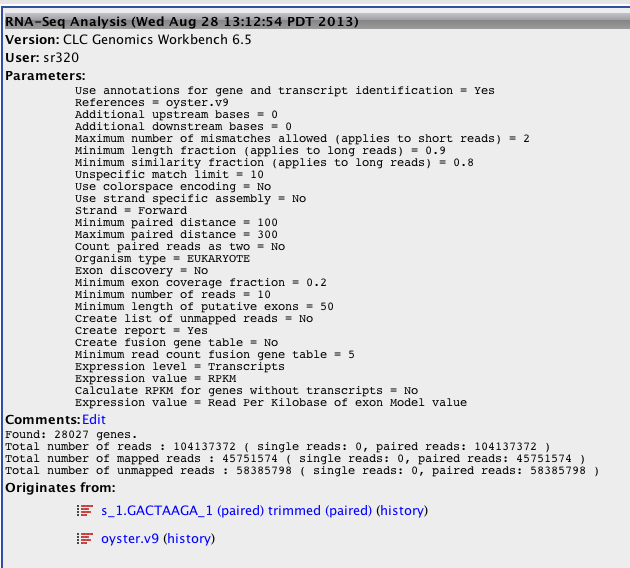
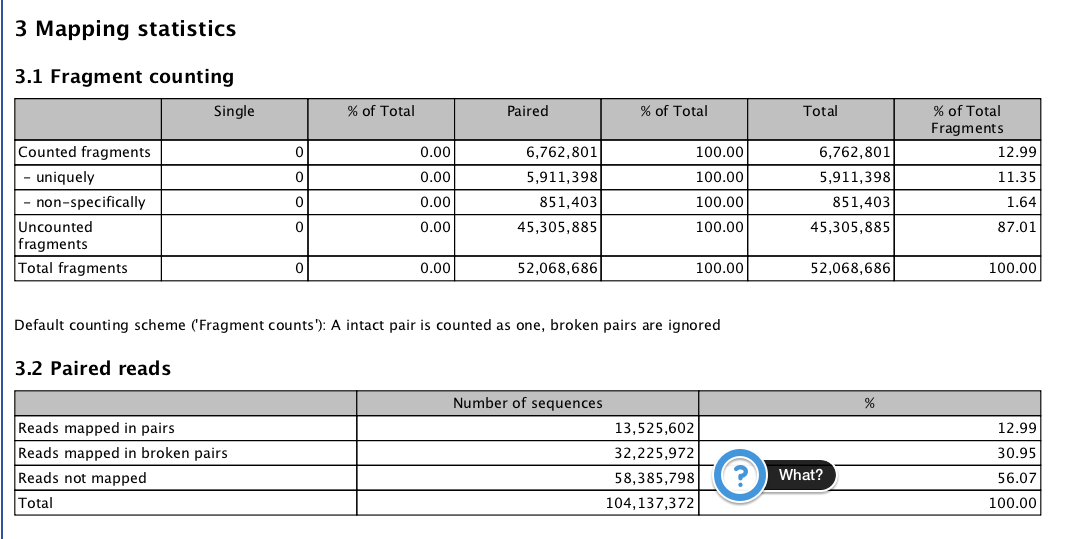
CLC Exon Specific Expression¶
#complete, though oddly not many reads map.....

CLC Gene Expression¶
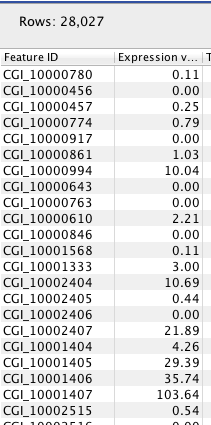
RPKM per Gene¶
#snapshot
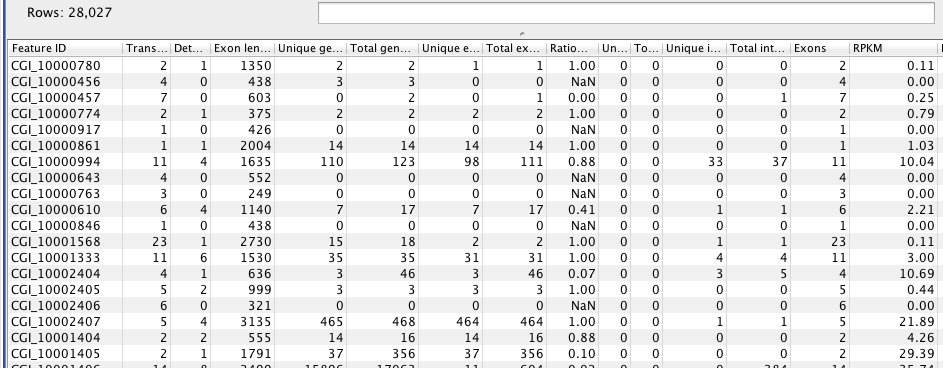
from pandas import *
# read data from data file into a pandas DataFrame
BiGillrna = read_table("http://eagle.fish.washington.edu/cnidarian/BiGill_RNAseq_gene.1" # name of the data file
#sep=",", # what character separates each column?
#na_values=["", " "]) # what values should be considered "blank" values?
)
BiGillrna
<class 'pandas.core.frame.DataFrame'> Int64Index: 28027 entries, 0 to 28026 Data columns: Feature ID 28027 non-null values Expression values 28027 non-null values Transcripts annotated 28027 non-null values Detected transcripts 28027 non-null values Exon length 28027 non-null values Unique gene reads 28027 non-null values Total gene reads 28027 non-null values Unique exon reads 28027 non-null values Total exon reads 28027 non-null values Ratio of unique to total (exon reads) 28027 non-null values Unique exon-exon reads 28027 non-null values Total exon-exon reads 28027 non-null values Unique intron-exon reads 28027 non-null values Total intron-exon reads 28027 non-null values Exons 28027 non-null values RPKM 28027 non-null values Median coverage 28027 non-null values Chromosome 28027 non-null values Chromosome region start 28027 non-null values Chromosome region end 28027 non-null values dtypes: float64(3), int64(14), object(3)
BiGillrna['Total gene reads'].hist(bins=1000);
#Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function.
#plt.axis([0, 0, 0, 20000])
#plt.xlabel('x', fontsize=20)
#plt.ylabel('RPKM', fontsize= 20)
#plt.title('Gill', fontsize= 20)
<matplotlib.axes.AxesSubplot at 0x10c2af390>

"""
Simple demo of a scatter plot.
"""
import numpy as np
import matplotlib.pyplot as plt
N = 50
x = BiGillrna['Total gene reads']
y = np.random.rand(N)
area = np.pi * (15 * np.random.rand(N))**2 # 0 to 15 point radiuses
plt.scatter(x, y, s=area, alpha=0.5)
plt.show()
--------------------------------------------------------------------------- ValueError Traceback (most recent call last) <ipython-input-30-d3694b00e2ae> in <module>() 11 area = np.pi * (15 * np.random.rand(N))**2 # 0 to 15 point radiuses 12 ---> 13 plt.scatter(x, y, s=area, alpha=0.5) 14 plt.show() /Users/sr320/anaconda/lib/python2.7/site-packages/matplotlib/pyplot.pyc in scatter(x, y, s, c, marker, cmap, norm, vmin, vmax, alpha, linewidths, faceted, verts, hold, **kwargs) 2916 vmin=vmin, vmax=vmax, alpha=alpha, 2917 linewidths=linewidths, faceted=faceted, verts=verts, -> 2918 **kwargs) 2919 draw_if_interactive() 2920 finally: /Users/sr320/anaconda/lib/python2.7/site-packages/matplotlib/axes.pyc in scatter(self, x, y, s, c, marker, cmap, norm, vmin, vmax, alpha, linewidths, faceted, verts, **kwargs) 6057 y = np.ma.ravel(y) 6058 if x.size != y.size: -> 6059 raise ValueError("x and y must be the same size") 6060 6061 s = np.ma.ravel(s) # This doesn't have to match x, y in size. ValueError: x and y must be the same size

File "<ipython-input-29-b7e33849ddba>", line 1 plot BiGillrna[Total gene reads] ^ SyntaxError: invalid syntax
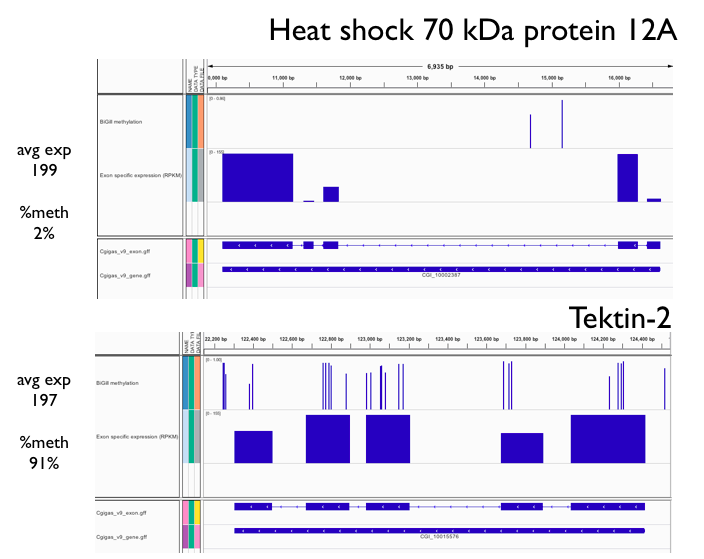
Identifying examples of MERV¶
Step 1) Based on exon specific expression data (RPKM) from CLC, identified all genes where number of exons was greater than 4 and exon expression was > 0 for all exons. This resulted in 38 genes.
Step 2) Calculated Expression Coefficient of Variance (stdev/mean RPKM) and average RPKM for all genes.
Step 3) For comparison, simply took the two genes with highest avg expression that also had a contrasting CV (1.6 v 0.5) AND similar avg expression levels (199 v 197) AND same number of exons (5).
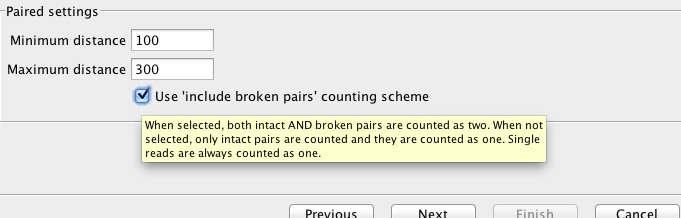
Changing Insert Size¶
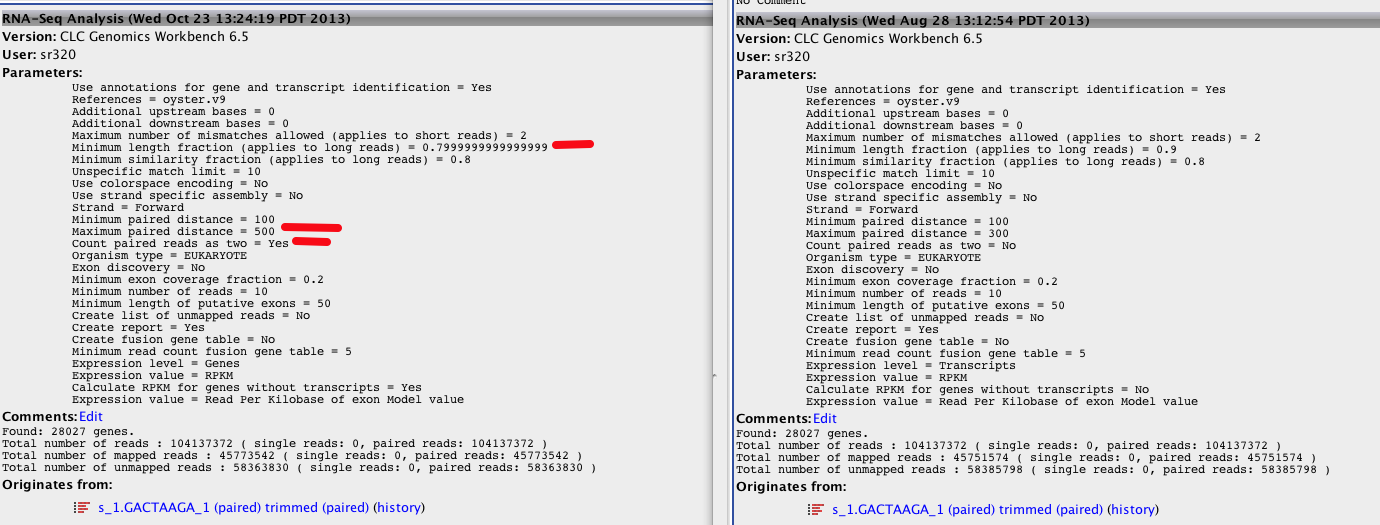
Upon changing insert size and inadvertably a couple more parameters, output changed
New (larger insert +)
Original