DaCosta & Sorenson (2016) single-end dd-RAD data set¶
Here we demonstrate a denovo assembly for an empirical RAD data set using the ipyrad Python API. This example was run on a workstation with 20 cores available and takes about <10 minutes to run completely, but can be run on even a laptop in about less than an hour.
We will use the Lagonosticta and Vidua data set from DaCosta & Sorenson 2016. This data set is composed of single-end 101bp reads from a ddRAD-seq library prepared with the SbfI and EcoRI enzymes and is available on NCBI by its study accession SRP059199. At the end of this notebook we also demonstrate the use of ipyrad.analysis tools to run downstream analyses on this data set.
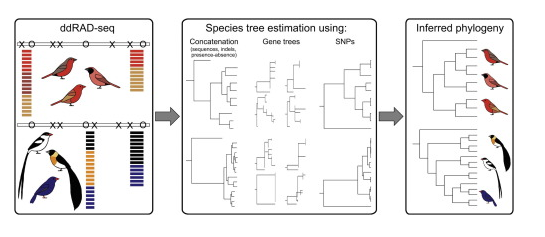
The figure below from this paper shows the general workflow in which two fairly distinct clades were sequenced together but then analyzed separately.
Setup (software and data files)¶
If you haven't done so yet, start by installing ipyrad using conda (see ipyrad installation instructions) as well as the packages in the cell below. This is easiest to do in a terminal. Then open a jupyter-notebook, like this one, and follow along with the tutorial by copying and executing the code in the cells, and adding your own documentation between them using markdown. Feel free to modify parameters to see their effects on the downstream results.
## conda install ipyrad -c ipyrad
## conda install toytree -c eaton-lab
## conda install entrez-direct -c bioconda
## conda install sratools -c bioconda
## imports
import ipyrad as ip
import ipyrad.analysis as ipa
import ipyparallel as ipp
In contrast to the ipyrad CLI, the ipyrad API gives users much more fine-scale control over the parallelization of their analysis, but this also requires learning a little bit about the library that we use to do this, called ipyparallel. This library is designed for use with jupyter-notebooks to allow massive-scale multi-processing while working interactively.
Understanding the nuts and bolts of it might take a little while, but it is fairly easy to get started using it, especially in the way it is integrated with ipyrad. To start a parallel client to you must run the command-line program 'ipcluster'. This will essentially start a number of independent Python processes (kernels) which we can then send bits of work to do. The cluster can be stopped and restarted independently of this notebook, which is convenient for working on a cluster where connecting to many cores is not always immediately available.
Open a terminal and type the following command to start an ipcluster instance with N engines.
## ipcluster start --n=20
## connect to cluster
ipyclient = ipp.Client()
ipyclient.ids
[0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19]
Download the data set (Finches)¶
These data are archived on the NCBI sequence read archive (SRA) under accession id SRP059199. For convenience, the data are also hosted at a public Dropbox link which is a bit easier to access. Run the code below to download and decompress the fastq data files, which will save them into a directory called fastqs-Finches/.
## download the Pedicularis data set from NCBI
sra = ipa.sratools(accession="SRP059199", workdir="fastqs-Finches")
sra.run(force=True, ipyclient=ipyclient)
[####################] 100% Downloading fastq files | 0:00:52 | 24 fastq files downloaded to /home/deren/Documents/ipyrad/tests/fastqs-Finches
Create an Assembly object¶
This object stores the parameters of the assembly and the organization of data files.
## you must provide a name for the Assembly
data = ip.Assembly("Finches")
New Assembly: Finches
Set parameters for the Assembly. This will raise an error if any of the parameters are not allowed because they are the wrong type, or out of the allowed range.
## set parameters
data.set_params("project_dir", "analysis-ipyrad/Finches")
data.set_params("sorted_fastq_path", "fastqs-Finches/*.fastq.gz")
data.set_params("datatype", "ddrad")
data.set_params("restriction_overhang", ("CCTGCAGG", "AATTC"))
data.set_params("clust_threshold", "0.85")
data.set_params("filter_adapters", "2")
data.set_params("max_Hs_consens", (5, 5))
data.set_params("output_formats", "psvnkua")
## see/print all parameters
data.get_params()
0 assembly_name Finches
1 project_dir ./analysis-ipyrad/Finches
2 raw_fastq_path
3 barcodes_path
4 sorted_fastq_path ./fastqs-Finches/*.fastq.gz
5 assembly_method denovo
6 reference_sequence
7 datatype ddrad
8 restriction_overhang ('CCTGCAGG', 'AATTC')
9 max_low_qual_bases 5
10 phred_Qscore_offset 33
11 mindepth_statistical 6
12 mindepth_majrule 6
13 maxdepth 10000
14 clust_threshold 0.85
15 max_barcode_mismatch 0
16 filter_adapters 2
17 filter_min_trim_len 35
18 max_alleles_consens 2
19 max_Ns_consens (5, 5)
20 max_Hs_consens (5, 5)
21 min_samples_locus 4
22 max_SNPs_locus (20, 20)
23 max_Indels_locus (8, 8)
24 max_shared_Hs_locus 0.5
25 trim_reads (0, 0, 0, 0)
26 trim_loci (0, 0, 0, 0)
27 output_formats ('p', 's', 'v', 'n', 'k', 'u')
28 pop_assign_file
Assemble the data set¶
## run steps 1 & 2 of the assembly
data.run("12")
Assembly: Finches [####################] 100% loading reads | 0:00:03 | s1 | [####################] 100% processing reads | 0:01:08 | s2 |
## access the stats of the assembly (so far) from the .stats attribute
data.stats
| state | reads_raw | reads_passed_filter | |
|---|---|---|---|
| Anomalospiza_imberbis | 2 | 935241 | 889028 |
| Clytospiza_monteiri | 2 | 1220879 | 1168704 |
| Lagonosticta_larvata | 2 | 1001743 | 944653 |
| Lagonosticta_rara | 2 | 992534 | 934386 |
| Lagonosticta_rhodopareia | 2 | 1020850 | 961277 |
| Lagonosticta_rubricata_congica | 2 | 1064587 | 997387 |
| Lagonosticta_rubricata_rubricata | 2 | 1079701 | 1009532 |
| Lagonosticta_rufopicta | 2 | 898117 | 846834 |
| Lagonosticta_sanguinodorsalis | 2 | 1034739 | 980499 |
| Lagonosticta_senegala_rendalli | 2 | 834688 | 786701 |
| Lagonosticta_senegala_rhodopsis | 2 | 792799 | 749508 |
| Vidua_chalybeata_amauropteryx | 2 | 1031674 | 1028936 |
| Vidua_chalybeata_neumanni | 2 | 709824 | 678382 |
| Vidua_fischeri | 2 | 998161 | 935631 |
| Vidua_hypocherina | 2 | 889818 | 844395 |
| Vidua_interjecta | 2 | 741675 | 708862 |
| Vidua_macroura_arenosa | 2 | 939649 | 891987 |
| Vidua_macroura_macroura | 2 | 729322 | 690496 |
| Vidua_obtusa | 2 | 809186 | 763098 |
| Vidua_orientalis | 2 | 862619 | 824073 |
| Vidua_paradisaea | 2 | 833981 | 791532 |
| Vidua_purpurascens | 2 | 927116 | 883478 |
| Vidua_raricola | 2 | 956686 | 907898 |
| Vidua_regia | 2 | 1012887 | 965657 |
## run steps 3-5 (within-sample steps) of the assembly
data.run("345")
Assembly: Finches [####################] 100% dereplicating | 0:00:03 | s3 | [####################] 100% clustering | 0:00:43 | s3 | [####################] 100% building clusters | 0:00:05 | s3 | [####################] 100% chunking | 0:00:01 | s3 | [####################] 100% aligning | 0:05:02 | s3 | [####################] 100% concatenating | 0:00:04 | s3 | [####################] 100% inferring [H, E] | 0:00:35 | s4 | [####################] 100% calculating depths | 0:00:03 | s5 | [####################] 100% chunking clusters | 0:00:02 | s5 | [####################] 100% consens calling | 0:01:24 | s5 |
Branch to create separate data sets for Vidua and Lagonistica¶
## create data set with only Vidua samples + outgroup
subs = [i for i in data.samples if "Vidua" in i] +\
[i for i in data.samples if "Anomalo" in i]
vidua = data.branch("vidua", subsamples=subs)
## create data set with only Lagonostica sampes + outgroup
subs = [i for i in data.samples if "Lagon" in i] +\
[i for i in data.samples if "Clyto" in i]
lagon = data.branch("lagon", subsamples=subs)
Assemble each data set through final steps¶
Or, if you are pressed for time, you can choose just one of the Assemblies going forward. If you do, you may want to choose Vidua since that is the one we use in the downstream analysis tools at the end of this notebook.
vidua.run("6")
Assembly: vidua [####################] 100% concat/shuffle input | 0:00:01 | s6 | [####################] 100% clustering across | 0:00:08 | s6 | [####################] 100% building clusters | 0:00:02 | s6 | [####################] 100% aligning clusters | 0:00:07 | s6 | [####################] 100% database indels | 0:00:03 | s6 | [####################] 100% indexing clusters | 0:00:01 | s6 | [####################] 100% building database | 0:00:04 | s6 |
lagon.run("6")
Assembly: lagon [####################] 100% concat/shuffle input | 0:00:01 | s6 | [####################] 100% clustering across | 0:00:06 | s6 | [####################] 100% building clusters | 0:00:01 | s6 | [####################] 100% aligning clusters | 0:00:04 | s6 | [####################] 100% database indels | 0:00:03 | s6 | [####################] 100% indexing clusters | 0:00:01 | s6 | [####################] 100% building database | 0:00:03 | s6 |
Branch to create several final data sets with different parameter settings¶
Here we use nested for-loops to iterate over assemblies and parameter values.
## iterate over data set and parameters
for assembly in [vidua, lagon]:
for min_sample in [4, 10]:
## create new assembly, apply new name and parameters
newname = "{}_min{}".format(assembly.name, min_sample)
newdata = assembly.branch(newname)
newdata.set_params("min_samples_locus", min_sample)
## run step 7
newdata.run("7")
Assembly: vidua_min4 [####################] 100% filtering loci | 0:00:09 | s7 | [####################] 100% building loci/stats | 0:00:01 | s7 | [####################] 100% building vcf file | 0:00:04 | s7 | [####################] 100% writing vcf file | 0:00:00 | s7 | [####################] 100% building arrays | 0:00:07 | s7 | [####################] 100% writing outfiles | 0:00:01 | s7 | Outfiles written to: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/vidua_min4_outfiles Assembly: vidua_min10 [####################] 100% filtering loci | 0:00:02 | s7 | [####################] 100% building loci/stats | 0:00:02 | s7 | [####################] 100% building vcf file | 0:00:02 | s7 | [####################] 100% writing vcf file | 0:00:00 | s7 | [####################] 100% building arrays | 0:00:07 | s7 | [####################] 100% writing outfiles | 0:00:01 | s7 | Outfiles written to: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/vidua_min10_outfiles Assembly: lagon_min4 [####################] 100% filtering loci | 0:00:02 | s7 | [####################] 100% building loci/stats | 0:00:01 | s7 | [####################] 100% building vcf file | 0:00:01 | s7 | [####################] 100% writing vcf file | 0:00:00 | s7 | [####################] 100% building arrays | 0:00:06 | s7 | [####################] 100% writing outfiles | 0:00:01 | s7 | Outfiles written to: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/lagon_min4_outfiles Assembly: lagon_min10 [####################] 100% filtering loci | 0:00:02 | s7 | [####################] 100% building loci/stats | 0:00:01 | s7 | [####################] 100% building vcf file | 0:00:02 | s7 | [####################] 100% writing vcf file | 0:00:00 | s7 | [####################] 100% building arrays | 0:00:06 | s7 | [####################] 100% writing outfiles | 0:00:01 | s7 | Outfiles written to: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/lagon_min10_outfiles
View final stats¶
The .stats attribute shows a stats summary for each sample, and a number of stats dataframes can be accessed for each step from the .stats_dfs attribute of the Assembly.
vm4 = ip.load_json("analysis-ipyrad/Finches/vidua_min4.json")
vm4.stats
loading Assembly: vidua_min4 from saved path: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/vidua_min4.json
| state | reads_raw | reads_passed_filter | clusters_total | clusters_hidepth | hetero_est | error_est | reads_consens | |
|---|---|---|---|---|---|---|---|---|
| Anomalospiza_imberbis | 6 | 935241 | 889028 | 19116 | 8770 | 0.007562 | 0.003823 | 8584 |
| Vidua_chalybeata_amauropteryx | 6 | 1031674 | 1028936 | 19674 | 8620 | 0.003011 | 0.000674 | 8535 |
| Vidua_chalybeata_neumanni | 6 | 709824 | 678382 | 22053 | 9035 | 0.005203 | 0.003346 | 8879 |
| Vidua_fischeri | 6 | 998161 | 935631 | 26309 | 9733 | 0.004910 | 0.002578 | 9595 |
| Vidua_hypocherina | 6 | 889818 | 844395 | 23806 | 9539 | 0.004199 | 0.002698 | 9406 |
| Vidua_interjecta | 6 | 741675 | 708862 | 22383 | 9345 | 0.006617 | 0.002864 | 9180 |
| Vidua_macroura_arenosa | 6 | 939649 | 891987 | 26536 | 10385 | 0.009633 | 0.003643 | 9994 |
| Vidua_macroura_macroura | 6 | 729322 | 690496 | 22449 | 9519 | 0.007330 | 0.002658 | 9350 |
| Vidua_obtusa | 6 | 809186 | 763098 | 26641 | 10039 | 0.006043 | 0.003828 | 9834 |
| Vidua_orientalis | 6 | 862619 | 824073 | 25127 | 9821 | 0.006461 | 0.002730 | 9666 |
| Vidua_paradisaea | 6 | 833981 | 791532 | 26286 | 9636 | 0.006648 | 0.004084 | 9360 |
| Vidua_purpurascens | 6 | 927116 | 883478 | 20212 | 9335 | 0.004603 | 0.002489 | 9196 |
| Vidua_raricola | 6 | 956686 | 907898 | 27028 | 10303 | 0.006500 | 0.002545 | 10115 |
| Vidua_regia | 6 | 1012887 | 965657 | 26355 | 9798 | 0.004225 | 0.002482 | 9657 |
lm4 = ip.load_json("analysis-ipyrad/Finches/lagon_min4.json")
lm4.stats
loading Assembly: lagon_min4 from saved path: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/lagon_min4.json
| state | reads_raw | reads_passed_filter | clusters_total | clusters_hidepth | hetero_est | error_est | reads_consens | |
|---|---|---|---|---|---|---|---|---|
| Clytospiza_monteiri | 6 | 1220879 | 1168704 | 32301 | 10216 | 0.008451 | 0.003611 | 9775 |
| Lagonosticta_larvata | 6 | 1001743 | 944653 | 25409 | 10557 | 0.008231 | 0.002764 | 10378 |
| Lagonosticta_rara | 6 | 992534 | 934386 | 29094 | 10016 | 0.008781 | 0.002809 | 9799 |
| Lagonosticta_rhodopareia | 6 | 1020850 | 961277 | 27767 | 9947 | 0.008752 | 0.003191 | 9714 |
| Lagonosticta_rubricata_congica | 6 | 1064587 | 997387 | 26398 | 10138 | 0.010114 | 0.003007 | 9887 |
| Lagonosticta_rubricata_rubricata | 6 | 1079701 | 1009532 | 22306 | 8723 | 0.008899 | 0.003063 | 8456 |
| Lagonosticta_rufopicta | 6 | 898117 | 846834 | 25233 | 10023 | 0.006609 | 0.003789 | 9802 |
| Lagonosticta_sanguinodorsalis | 6 | 1034739 | 980499 | 27716 | 9046 | 0.009435 | 0.004015 | 8677 |
| Lagonosticta_senegala_rendalli | 6 | 834688 | 786701 | 21363 | 9960 | 0.007350 | 0.002776 | 9747 |
| Lagonosticta_senegala_rhodopsis | 6 | 792799 | 749508 | 26921 | 9379 | 0.006013 | 0.003181 | 9194 |
## or read the full stats file as a bash command (cat)
!cat $vm4.stats_files.s7
## The number of loci caught by each filter.
## ipyrad API location: [assembly].stats_dfs.s7_filters
total_filters applied_order retained_loci
total_prefiltered_loci 13949 0 13949
filtered_by_rm_duplicates 1645 1645 12304
filtered_by_max_indels 233 233 12071
filtered_by_max_snps 268 9 12062
filtered_by_max_shared_het 83 55 12007
filtered_by_min_sample 3583 3310 8697
filtered_by_max_alleles 724 251 8446
total_filtered_loci 8446 0 8446
## The number of loci recovered for each Sample.
## ipyrad API location: [assembly].stats_dfs.s7_samples
sample_coverage
Anomalospiza_imberbis 3921
Vidua_chalybeata_amauropteryx 5701
Vidua_chalybeata_neumanni 6548
Vidua_fischeri 6296
Vidua_hypocherina 6377
Vidua_interjecta 6590
Vidua_macroura_arenosa 6548
Vidua_macroura_macroura 6396
Vidua_obtusa 6898
Vidua_orientalis 6746
Vidua_paradisaea 6699
Vidua_purpurascens 6674
Vidua_raricola 6991
Vidua_regia 6267
## The number of loci for which N taxa have data.
## ipyrad API location: [assembly].stats_dfs.s7_loci
locus_coverage sum_coverage
1 0 0
2 0 0
3 0 0
4 915 915
5 480 1395
6 444 1839
7 334 2173
8 368 2541
9 348 2889
10 400 3289
11 423 3712
12 760 4472
13 1895 6367
14 2079 8446
## The distribution of SNPs (var and pis) per locus.
## var = Number of loci with n variable sites (pis + autapomorphies)
## pis = Number of loci with n parsimony informative site (minor allele in >1 sample)
## ipyrad API location: [assembly].stats_dfs.s7_snps
var sum_var pis sum_pis
0 941 0 3168 0
1 1169 1169 2220 2220
2 1144 3457 1454 5128
3 1096 6745 813 7567
4 855 10165 384 9103
5 723 13780 200 10103
6 629 17554 94 10667
7 516 21166 63 11108
8 391 24294 26 11316
9 293 26931 9 11397
10 221 29141 7 11467
11 151 30802 3 11500
12 115 32182 2 11524
13 74 33144 1 11537
14 46 33788 2 11565
15 34 34298 0 11565
16 20 34618 0 11565
17 11 34805 0 11565
18 11 35003 0 11565
19 5 35098 0 11565
20 1 35118 0 11565
## Final Sample stats summary
state reads_raw reads_passed_filter clusters_total clusters_hidepth hetero_est error_est reads_consens loci_in_assembly
Anomalospiza_imberbis 7 935241 889028 19116 8770 0.008 3.823e-03 8584 3921
Vidua_chalybeata_amauropteryx 7 1031674 1028936 19674 8620 0.003 6.740e-04 8535 5701
Vidua_chalybeata_neumanni 7 709824 678382 22053 9035 0.005 3.346e-03 8879 6548
Vidua_fischeri 7 998161 935631 26309 9733 0.005 2.578e-03 9595 6296
Vidua_hypocherina 7 889818 844395 23806 9539 0.004 2.698e-03 9406 6377
Vidua_interjecta 7 741675 708862 22383 9345 0.007 2.864e-03 9180 6590
Vidua_macroura_arenosa 7 939649 891987 26536 10385 0.010 3.643e-03 9994 6548
Vidua_macroura_macroura 7 729322 690496 22449 9519 0.007 2.658e-03 9350 6396
Vidua_obtusa 7 809186 763098 26641 10039 0.006 3.828e-03 9834 6898
Vidua_orientalis 7 862619 824073 25127 9821 0.006 2.730e-03 9666 6746
Vidua_paradisaea 7 833981 791532 26286 9636 0.007 4.084e-03 9360 6699
Vidua_purpurascens 7 927116 883478 20212 9335 0.005 2.489e-03 9196 6674
Vidua_raricola 7 956686 907898 27028 10303 0.007 2.545e-03 10115 6991
Vidua_regia 7 1012887 965657 26355 9798 0.004 2.482e-03 9657 6267
## the same full stats for lagon
!cat $lm4.stats_files.s7
## The number of loci caught by each filter.
## ipyrad API location: [assembly].stats_dfs.s7_filters
total_filters applied_order retained_loci
total_prefiltered_loci 14533 0 14533
filtered_by_rm_duplicates 1717 1717 12816
filtered_by_max_indels 282 282 12534
filtered_by_max_snps 264 14 12520
filtered_by_max_shared_het 84 65 12455
filtered_by_min_sample 5209 4689 7766
filtered_by_max_alleles 797 268 7498
total_filtered_loci 7498 0 7498
## The number of loci recovered for each Sample.
## ipyrad API location: [assembly].stats_dfs.s7_samples
sample_coverage
Clytospiza_monteiri 4776
Lagonosticta_larvata 6057
Lagonosticta_rara 5737
Lagonosticta_rhodopareia 6061
Lagonosticta_rubricata_congica 6183
Lagonosticta_rubricata_rubricata 4741
Lagonosticta_rufopicta 5500
Lagonosticta_sanguinodorsalis 5512
Lagonosticta_senegala_rendalli 5491
Lagonosticta_senegala_rhodopsis 5309
## The number of loci for which N taxa have data.
## ipyrad API location: [assembly].stats_dfs.s7_loci
locus_coverage sum_coverage
1 0 0
2 0 0
3 0 0
4 1141 1141
5 794 1935
6 840 2775
7 752 3527
8 911 4438
9 1359 5797
10 1701 7498
## The distribution of SNPs (var and pis) per locus.
## var = Number of loci with n variable sites (pis + autapomorphies)
## pis = Number of loci with n parsimony informative site (minor allele in >1 sample)
## ipyrad API location: [assembly].stats_dfs.s7_snps
var sum_var pis sum_pis
0 313 0 2301 0
1 640 640 1969 1969
2 806 2252 1296 4561
3 895 4937 835 7066
4 812 8185 521 9150
5 808 12225 255 10425
6 705 16455 151 11331
7 617 20774 73 11842
8 516 24902 41 12170
9 375 28277 25 12395
10 277 31047 16 12555
11 196 33203 8 12643
12 197 35567 3 12679
13 119 37114 2 12705
14 84 38290 0 12705
15 54 39100 1 12720
16 42 39772 1 12736
17 17 40061 0 12736
18 16 40349 0 12736
19 3 40406 0 12736
20 6 40526 0 12736
## Final Sample stats summary
state reads_raw reads_passed_filter clusters_total clusters_hidepth hetero_est error_est reads_consens loci_in_assembly
Clytospiza_monteiri 7 1220879 1168704 32301 10216 0.008 0.004 9775 4776
Lagonosticta_larvata 7 1001743 944653 25409 10557 0.008 0.003 10378 6057
Lagonosticta_rara 7 992534 934386 29094 10016 0.009 0.003 9799 5737
Lagonosticta_rhodopareia 7 1020850 961277 27767 9947 0.009 0.003 9714 6061
Lagonosticta_rubricata_congica 7 1064587 997387 26398 10138 0.010 0.003 9887 6183
Lagonosticta_rubricata_rubricata 7 1079701 1009532 22306 8723 0.009 0.003 8456 4741
Lagonosticta_rufopicta 7 898117 846834 25233 10023 0.007 0.004 9802 5500
Lagonosticta_sanguinodorsalis 7 1034739 980499 27716 9046 0.009 0.004 8677 5512
Lagonosticta_senegala_rendalli 7 834688 786701 21363 9960 0.007 0.003 9747 5491
Lagonosticta_senegala_rhodopsis 7 792799 749508 26921 9379 0.006 0.003 9194 5309
Analysis tools¶
Thee is a lot more information about analysis tools in the ipyrad documentation. But here I'll show just a quick example of how you can easily access the data files for these assemblies and use them in downstream analysis software. The ipyrad analysis tools include convenient wrappers to make it easier to parallelize analyses of RAD-seq data. You should still read the full tutorial of the software you are using to understand the full scope of the parameters involved and their impacts, but once you understand that, the ipyrad analysis tools provide an easy way to setup up scripts to sample different distributions of SNPs and to run many replicates in parallel.
import ipyrad.analysis as ipa
## you can re-load assemblies at a later time from their JSON file
min4 = ip.load_json("analysis-ipyrad/Finches/vidua_min4.json")
min10 = ip.load_json("analysis-ipyrad/Finches/vidua_min10.json")
loading Assembly: vidua_min4 from saved path: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/vidua_min4.json loading Assembly: vidua_min10 from saved path: ~/Documents/ipyrad/tests/analysis-ipyrad/Finches/vidua_min10.json
RAxML -- ML concatenation tree inference¶
## conda install raxml -c bioconda
## conda install toytree -c eaton-lab
## create a raxml analysis object for the min13 data sets
rax = ipa.raxml(
name=min10.name,
data=min10.outfiles.phy,
workdir="analysis-raxml",
T=20,
N=100,
o=[i for i in min10.samples if "Ano" in i],
)
## print the raxml command and call it
print rax.command
rax.run(force=True)
raxmlHPC-PTHREADS-SSE3 -f a -T 20 -m GTRGAMMA -N 100 -x 12345 -p 54321 -n vidua_min10 -w /home/deren/Documents/ipyrad/tests/analysis-raxml -s /home/deren/Documents/ipyrad/tests/analysis-ipyrad/Finches/vidua_min10_outfiles/vidua_min10.phy -o Anomalospiza_imberbis job vidua_min10 finished successfully
## access the resulting tree files
rax.trees
bestTree ~/Documents/ipyrad/tests/analysis-raxml/RAxML_bestTree.vidua_min10 bipartitions ~/Documents/ipyrad/tests/analysis-raxml/RAxML_bipartitions.vidua_min10 bipartitionsBranchLabels ~/Documents/ipyrad/tests/analysis-raxml/RAxML_bipartitionsBranchLabels.vidua_min10 bootstrap ~/Documents/ipyrad/tests/analysis-raxml/RAxML_bootstrap.vidua_min10 info ~/Documents/ipyrad/tests/analysis-raxml/RAxML_info.vidua_min10
## plot a tree in the notebook with toytree
import toytree
tre = toytree.tree(rax.trees.bipartitions)
tre.root(wildcard="Ano")
tre.draw(
width=350,
height=400,
node_labels=tre.get_node_values("support"),
#use_edge_lengths=True,
);
tetrad -- quartet tree inference¶
## create a tetrad analysis object
tet = ipa.tetrad(
name=min4.name,
seqfile=min4.outfiles.snpsphy,
mapfile=min4.outfiles.snpsmap,
nboots=100,
)
loading seq array [14 taxa x 35118 bp] max unlinked SNPs per quartet (nloci): 7505
## run tree inference
tet.run(ipyclient)
host compute node: [40 cores] on tinus inferring 1001 induced quartet trees [####################] 100% initial tree | 0:00:04 | [####################] 100% boot 100 | 0:01:35 |
## access tree files
tet.trees
boots ~/Documents/ipyrad/tests/analysis-tetrad/vidua_min4.boots cons ~/Documents/ipyrad/tests/analysis-tetrad/vidua_min4.cons nhx ~/Documents/ipyrad/tests/analysis-tetrad/vidua_min4.nhx tree ~/Documents/ipyrad/tests/analysis-tetrad/vidua_min4.tree
## plot results (just like above, but unrooted by default)
import toytree
qtre = toytree.tree(tet.trees.nhx)
qtre.root(wildcard="Ano")
qtre.draw(
width=350,
height=400,
node_labels=tre.get_node_values("support"),
);
## draw a cloud-tree to see variation among bootstrap trees
## note that the trees are UNROOTED here, but tips are in the
## same order in all trees.
boots = toytree.multitree(tet.trees.boots, fixed_order=tre.get_tip_labels())
boots.draw_cloudtree(orient='right', edge_style={"opacity": 0.05});
STRUCTURE -- population cluster inference¶
## conda install structure clumpp -c ipyrad
## create a structure analysis object for the no-outgroup data set
struct = ipa.structure(
name=min10.name,
data=min10.outfiles.str,
mapfile=min10.outfiles.snpsmap,
)
## set params for analysis (should be longer in real analyses)
struct.mainparams.burnin=1000
struct.mainparams.numreps=8000
## run structure across 10 random replicates of sampled unlinked SNPs
for kpop in [2, 4, 6, 8]:
struct.run(kpop=kpop, nreps=10, ipyclient=ipyclient)
submitted 10 structure jobs [vidua_min10-K-2] submitted 10 structure jobs [vidua_min10-K-4] submitted 10 structure jobs [vidua_min10-K-6] submitted 10 structure jobs [vidua_min10-K-8]
## wait for all of these jobs to finish
ipyclient.wait()
True
Use Clumpp to permute replicates¶
## these options make it run faster
struct.clumppparams.m = 3 ## use largegreedy algorithm
struct.clumppparams.greedy_option = 2 ## test nrepeat possible orders
struct.clumppparams.repeats = 10000 ## number of repeats
## collect results
tables = {}
for kpop in [2, 4, 6, 8]:
tables[kpop] = struct.get_clumpp_table(kpop)
mean scores across 10 replicates. mean scores across 10 replicates. mean scores across 10 replicates. mean scores across 10 replicates.
Plot the results as a barplot¶
Usually a next step in a structure analysis would be do some kind of statistical analysis to compare models and identify K values that fit the data well.
## order of bars will be taken from ladderized tree above
myorder = tre.get_tip_labels()
## import toyplot (packaged with toytree)
import toyplot
## plot bars for each K-value (mean of 10 reps)
for kpop in [2, 4, 6, 8]:
table = tables[kpop]
table = table.ix[myorder]
## plot barplot w/ hover
canvas, axes, mark = toyplot.bars(
table,
title=[[i] for i in table.index.tolist()],
width=400,
height=200,
yshow=False,
style={"stroke": toyplot.color.near_black},
)
TREEMIX -- ML tree & admixture co-inference¶
## conda install treemix -c ipyrad
## group taxa into 'populations'
imap = {
'orient': ['Vidua_orientalis'],
'interj': ['Vidua_interjecta'],
'obtusa': ['Vidua_obtusa'],
'paradi': ['Vidua_paradisaea'],
'hypoch': ['Vidua_hypocherina'],
'macrou': ['Vidua_macroura_macroura', 'Vidua_macroura_arenosa'],
'fische': ['Vidua_fischeri'],
'regia' : ['Vidua_regia'],
'chalyb': ['Vidua_chalybeata_amauropteryx', 'Vidua_chalybeata_neumanni'],
'purpur': ['Vidua_purpurascens'],
'rarico': ['Vidua_raricola'],
#'outgro': ['Anomalospiza_imberbis'],
}
## optional: loci will be filtered if they do not have data for at
## least N samples in each species. Minimums cannot be <1.
minmap = {
'orient': 1,
'interj': 1,
'obtusa': 1,
'paradi': 1,
'hypoch': 1,
'macrou': 2,
'fische': 1,
'regia' : 1,
'chalyb': 2,
'purpur': 1,
'rarico': 1,
#'outgro': 1,
}
## create a treemix analysis object
tmix = ipa.treemix(
name=min10.name,
data=min10.outfiles.snpsphy,
imap=imap,
minmap=minmap,
)
## set params on treemix object
tmix.params.m = 1
tmix.params.root = "interj,orient,paradi,obtusa"
tmix.params.global_ = 1
## you can simply write the input files and run them externally
## or, as we show below, use the .run() command to run them here.
tmix.write_output_file()
ntaxa 14; nSNPs total 35118; nSNPs written 16364
## a dictionary for storing treemix objects
tdict = {}
## iterate over values of m
for rep in xrange(4):
for mig in xrange(4):
## create new treemix object copy
name = "mig-{}-rep-{}".format(mig, rep)
tmp = tmix.copy(name)
## set params on new object
tmp.params.m = mig
## run treemix analysis
tmp.run()
## store the treemix object
tdict[name] = tmp
import toyplot
import numpy as np
canvas = toyplot.Canvas(width=800, height=1200)
idx = 0
for mig in range(4):
for rep in range(4):
tmp = tdict["mig-{}-rep-{}".format(mig, rep)]
ax = canvas.cartesian(grid=(4, 4, idx), padding=25, margin=(25, 50, 100, 25))
ax = tmp.draw(ax)
idx += 1
baba: D-statistic (ABBA-BABA) admixture inference/hypothesis testing¶
## create a baba analysis object
bb = ipa.baba(
data=min4.outfiles.loci,
newick="analysis-raxml/RAxML_bestTree.vidua_min10"
)
## this will generate tests from the tree, using constraints.
bb.generate_tests_from_tree(
constraint_exact=False,
constraint_dict={
"p4": ['Anomalospiza_imberbis'],
'p3': ['Vidua_macroura_macroura', 'Vidua_macroura_arenosa'],
})
168 tests generated from tree
## run inference and significance testing on tests
bb.run(ipyclient=ipyclient)
[####################] 100% calculating D-stats | 0:00:24 |
## sorted results for the tests performed
bb.results_table.sort_values(by="Z", ascending=False)
| dstat | bootmean | bootstd | Z | ABBA | BABA | nloci | |
|---|---|---|---|---|---|---|---|
| 25 | -0.195 | -1.952e-01 | 0.071 | 2.751 | 43.250 | 64.250 | 3207 |
| 22 | 0.212 | 2.116e-01 | 0.078 | 2.729 | 61.000 | 39.625 | 3143 |
| 1 | -0.186 | -1.816e-01 | 0.069 | 2.701 | 44.656 | 65.094 | 3262 |
| 13 | -0.192 | -1.888e-01 | 0.074 | 2.608 | 41.875 | 61.750 | 3186 |
| 15 | -0.212 | -2.081e-01 | 0.084 | 2.528 | 39.625 | 61.000 | 3143 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 120 | -0.009 | -2.353e-03 | 0.104 | 0.085 | 34.750 | 35.375 | 2717 |
| 157 | 0.004 | 5.400e-03 | 0.085 | 0.047 | 46.812 | 46.438 | 3222 |
| 0 | 0.002 | 4.923e-03 | 0.071 | 0.030 | 58.312 | 58.062 | 3268 |
| 76 | -0.003 | -6.335e-04 | 0.094 | 0.027 | 36.500 | 36.688 | 2765 |
| 70 | 0.003 | 1.563e-03 | 0.095 | 0.027 | 36.688 | 36.500 | 2765 |
168 rows × 7 columns
summary¶
The top results show greatest support for admixture between V. macroura and V. paradisaea, though the signal is not very strong (Z=2.7).
## the test that had the most significant result: (BABA)
bb.tests[25]
{'p1': ['Vidua_paradisaea'],
'p2': ['Vidua_orientalis', 'Vidua_interjecta'],
'p3': ['Vidua_macroura_arenosa'],
'p4': ['Anomalospiza_imberbis']}
## next best (ABBA)
bb.tests[22]
{'p1': ['Vidua_orientalis'],
'p2': ['Vidua_paradisaea'],
'p3': ['Vidua_macroura_macroura'],
'p4': ['Anomalospiza_imberbis']}