Previously on Py4Life¶
- Modules
- Files
- Regex
- Debugging
In today's episode¶
- Biopython
- Sequence analysis
- Sequence data in python
Biopython¶
What is Biopython?¶
Biopython is a set of freely available tools for biological computation written in Python by an international team of developers.
(from the Biopython official website).
Simply put, Biopython is a python package (collection of modules), which includes lots of functions and tools associated with bioinformatics.
What's in the Biopython package?¶
- Ready-to-use parsers, dedicated to biological data file formats:
- FASTA
- GenBank
- BLAST output
- Newick\Nexus phylogenetic trees
- Special data types to handle biological data:
- Nucleotide sequences
- Protein sequences
- Protein structure
- Phylogenetic trees
- Easy accessibility to biological databases:
- NCBI BLAST
- PubMed
- GenBank
- SwissProt
- Bioinformatic tools:
- MSA
- Clustering
- Sequence manipulation
And lots of other stuff...
We will not cover everything today.
Installing Biopython¶
Biopython is not part of the core distribution and thus needs to be installed before it can be used. To install the package, use pip install Biopython or another installation method, as explained in lecture 4.
Using Biopython¶
Once installed, we can import the package using:
import Bio
In practice, we won't usually do that, because this package is huge, and we only need certain parts of its functionality. Therefore, we can do:
from Bio import <module name>. For example:
from Bio import SeqIO
Documentation¶
Biopython has very good and very vast documentation.
A great tutorial is available here.
If you need a specific task, you might want to take a look at the 'Cookbook' pages or the wiki pages.
Biopython's online community is very active. Join one of the mailing lists for lots of useful discussions and lots of spam...
Introducing Sequence objects¶

So far in this course, we have seen most of the basic data types in Python: str,int,float,list,dict etc. We've also briefly talked about some specialized data types such as file objects (generated when using the open() function) and the csv reader.
One of the most basic objects in bioinformatics are sequences, and Biopython has a whole mechanism for dealing with sequences. This is done by introducing a new data type, namely a sequence data type.
In principal, we could just use strings to represent biological sequences.
seq = "AGTACACTGGT"
But using Biopython's specialized data type, we get much more functionality, novel to sequences, without giving away the string functionality. That's achieved thanks to special methods of the sequence data type (but not the string data type).
Creating sequence objects¶
To work with sequence objects, we'll first have to import the relevant class (specific part of the module):
from Bio.Seq import Seq
Now, creating a sequence object is done using Seq, on the required sequence string. Like this:
my_DNA_seq = Seq("AGTACACTGGT")
print(type(seq))
print(type(my_DNA_seq))
We can store amino acid sequences in the same way:
my_AA_seq = Seq("EVRNAK")
print(type(my_AA_seq))
What can we do with sequence objects?¶
As we said earlier, sequence objects have all the functionality that strings have. That means that everything that we've done with strings can also be done with sequence objects. Some examples:
# printing
print(my_DNA_seq)
# length
print(len(my_DNA_seq))
# Accessing specific elements
print(my_DNA_seq[0])
print(my_DNA_seq[7])
print(my_DNA_seq[-1])
# Slicing
print(my_DNA_seq[3:10])
Notice that slicing a sequence object results in a new sequence object:
partial_DNA = my_DNA_seq[3:10]
print(type(partial_DNA))
# Sequence concatenation
seq1 = Seq("AGGCACCATTA")
seq2 = Seq("TTACAGGA")
whole_seq = seq1 + seq2
print(whole_seq)
OK, so here's when it gets interesting: sequence objects have some special and powerful methods. For example, finding the complement and the reverse-complement has never been easier:
my_DNA_seq = Seq("GATCGATGGGCCTATATAGGATCGAAAATCGC")
complement_seq = my_DNA_seq.complement()
print(complement_seq)
rev_comp_seq = my_DNA_seq.reverse_complement()
print(rev_comp_seq)
We can transcribe DNA to RNA, which is basically just changing T -> U (assuming that we are using the 'sense' strand).
coding_DNA = Seq("ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG")
mRNA = coding_DNA.transcribe()
print(mRNA)
And also translate to protein sequence, either from RNA or straight from coding (sense) DNA:
peptide = mRNA.translate()
print(peptide)
Class exercise 6A¶
Write a function that receives antisense DNA, as a string and returns it's translation, as a sequence object. Test your code on the given string sequence (displayed 5' to 3').
from Bio.Seq import Seq
def antisense_string_to_protein_seq(DNA_string):
antisense_seq = Seq(DNA_string)
sense_seq = antisense_seq.reverse_complement()
prot_seq = sense_seq.translate()
return prot_seq
antisense_DNA = "TACCGGTAACATTACCCGGCGACTTTCCCACGGGCTATC"
protein = antisense_string_to_protein_seq(antisense_DNA)
print(protein)
assert str(protein) == 'DSPWESRRVMLPV'
assert isinstance(protein,Seq)
Advancing to sequence record objects¶
Representing sequences is great, but sometimes we want to associate more information with the sequence object, such as the gene name, references, organism and general descriptions. To do that, we move from simple sequence objects to more complex ones, called Sequence record objects.
GenBank¶
GenBank is a database of all publicly available nucleotide data collected by scientists, maintained by NCBI. Sequences in GenBank are annotated, meaning that the sequence itself is accompanied by additional data, regarding the meaning, source and other details of the sequence. Each sequence, along with its accompanying data, is called a record. As part of the NCBI collection of databases, GenBank can be searched using a dedicated search engine, called Entrez.
To better understand the idea of sequence records, lets take a look at the GenBank record for the MatK gene of Oryza sativa (rice).
from IPython.display import HTML
HTML('<iframe src=http://www.ncbi.nlm.nih.gov/nuccore/JN114766.1 width=1000 height=350></iframe>')
Notice that there are lots of additional data besides the sequence itself. So a sequence record object is basically just a sequence object, associaated to some other simple data fields.
Creating sequence record objects¶
In principal, we can create sequence record objects 'from scratch', like we did with simple sequence objects. However, in practice this is rarely used. Instead, we usually get sequence record objects from data parsers. For example, here we get the GenBank record shown above as a sequence record object.
from Bio import Entrez
from Bio import SeqIO
try:
handle = Entrez.efetch(db="nucleotide", rettype="gb", retmode="text", id="387865390")
O_sativa_matK = SeqIO.read(handle, "gb")
handle.close()
print(type(O_sativa_matK))
except:
print('Failed to fetch record.')
We'll soon learn to use the SeqIO module, but for now let's just look at what we can do with the object we got.
A sequence record object contains some data fields which we can extract by calling their names.
The most basic is the sequence itself, which is called using .seq:
sequence = O_sativa_matK.seq
print(sequence)
print(type(sequence))
Note that we got a sequence object, not just a string.
Some other basic data fields of the sequence record object are the id, name and description:
print(O_sativa_matK.id)
print(O_sativa_matK.name)
print(O_sativa_matK.description)
There are a couple of other data fields, but we will only look at one. The annotations field contains more general information about the sequence. In fact, it's a dictionary where the keys are the names of the information and the values are the actual information. For example:
# get the organism
print(O_sativa_matK.annotations['organism'])
# get the DNA source
print(O_sativa_matK.annotations['source'])
# get date of submission
print(O_sativa_matK.annotations['date'])
Another data field worth mentioning is the features field, describing the roles and locations of higher order sequence domains and elements within a record. This is especially relevant for records describing long sequences such as whole chromosomes. Record features are further discussed in HW6.
Sequence I/O¶
The SeqIO module is the easiest way to deal with sequences. It's a wrapper for the Seq and SeqRecord (and other) modules and lets us manage, manipulate, read and write sequence data without doing any parsing.
As usual, we'll start by importing the module:
from Bio import SeqIO
Reading sequence files¶
The file Orchids.fasta contains multiple DNA sequences of Orchids, stored in the FASTA format. In lesson 4 we wrote our own function for parsing fasta files, but the SeqIO module has its own parsers, which we'll learn how to use.
The most useful function within the SeqIO module is SeqIO.parse(). This function is used to read files including sequence data in one of the standard formats.
fasta_handle = SeqIO.parse('lec6_files/Orchids.fasta','fasta')
The function expects two parameters: a file path and a string specifying the file format (a full list of supported formats available here). It returns a special object which lets us read the file. Once we have the handle, we can use it to iterate over the sequence records in the file, using a simple for loop:
for seq_record in fasta_handle:
print(seq_record.id,len(seq_record))
As you can see, each iteration of the loop yields a SeqRecord object, which we can treat as we did in the previous section. For example, here we extracted the id and the length of each sequence.
Similarly, we can parse a GenBank format file with the same function. All we have to do is replace the 'fasta' string with 'genbank'. We'll also do it in a more elegant, one-step code:
for seq_record in SeqIO.parse('lec6_files/Orchids.gbk','genbank'):
print(seq_record.id,len(seq_record))
Another useful trick is converting the sequence file handle into a list, so its elements (SeqRecord objects) are more easily accessed:
orchid_records_list = list(SeqIO.parse('lec6_files/Orchids.gbk','genbank'))
# last record id
print(orchid_records_list[-1].id)
# number of records in file
print(len(orchid_records_list))
This is not a good idea though, when dealing with large files, since very long lists are not desired.
If your file contains only one record, you can use SeqIO.read() which will return a SeqRecord object.
record = SeqIO.read('lec6_files/P_emersonii.gbk', 'genbank')
print(record.seq)
Class exercise 6B¶
Write a function that reads a GenBank file and returns the unique species names found in the file. Use the .annotations field to get the organism name of each record.. The species is always the second word of the organism name. The set() command will ensure you get unique species (each species will appear only once). Complete the code below:
from Bio import SeqIO
def get_unique_species(gb_file):
species_list = []
# iterate on file records
for seq_record in SeqIO.parse(gb_file,'genbank'):
# get species
record_organism = seq_record.annotations['organism']
species = record_organism.split()[1] # get the second word
# insert species to list
species_list.append(species)
return set(species_list)
orchids_species = get_unique_species('lec6_files/Orchids.gbk')
print(orchids_species)
assert len(orchids_species) == 92
Reading sequence files as dictionaries¶
Another nice option for dealing with sequence files is reading them as dictionaries. This is done using SeqIO.to_dict() function. to_dict() takes a sequence file handle, created by SeqIO.parse(), as an argument.
handle = SeqIO.parse('lec6_files/Orchids.gbk','genbank')
orchids_dict = SeqIO.to_dict(handle)
The function returns a dictionary, which we can work with as usual.
print(orchids_dict.keys())
So the keys are the IDs, and the values are SeqRecord objects. It's very easy to access specif records this way:
print(orchids_dict['Z78481.1'].seq)
Again, this could get tricky when working with large files due to memory problems.
Writing sequence files¶
SeqIO also makes it easy to write sequence files. For that, we have the SeqIO.write() function. It receives a list of SeqRecords and writes them to an output file, in a format of your choice.
For example, the following function reads a GenBank file, and prints to a new file only records that belong to the organism specified by the user.
def filter_GB_file_by_organism(input_file,organism,output_file):
organism_records = []
# read the input file
for record in SeqIO.parse(input_file,'genbank'):
rec_organism = record.annotations['organism']
if rec_organism == organism:
organism_records.append(record)
# write the desired records to output
SeqIO.write(organism_records,output_file,'genbank') # notice the 3 arguments
orchids_file = 'lec6_files/Orchids.gbk'
C_irapeanum_file = 'lec6_files/C_irapeanum.gbk'
P_emersonii_file = 'lec6_files/P_emersonii.gbk'
filter_GB_file_by_organism(orchids_file,'Cypripedium irapeanum',C_irapeanum_file)
filter_GB_file_by_organism(orchids_file,'Paphiopedilum emersonii',P_emersonii_file)
We can easily change the function to create files in a different format. This format may be set by the user of the function:
def filter_GB_file_by_organism(input_file,organism,output_file, output_format = 'fasta'):
organism_records = []
# read the input file
for record in SeqIO.parse(input_file,'genbank'):
rec_organism = record.annotations['organism']
if rec_organism == organism:
organism_records.append(record)
# write the desired records to output
SeqIO.write(organism_records,output_file, output_format) # <--- changed to fasta here
Which means that SeqIO.write() can be used to convert between file formats. However, this is not necessary, since SeqIO has its built-in convert() function:
SeqIO.convert('lec6_files/P_emersonii.gbk','genbank','lec6_files/P_emersonii.fasta','fasta')
A BLAST from the past¶

BLAST - Basic Local Alignment and Search Tool, is one of the most important methods in bioinformatics.
Biopython lets you run BLAST from within your code, retrieve and analyze the results and save them. But before we see how to do that, lets recall how BLAST actually works.
BLAST - reminder¶
BLAST is an algorithm used for comparing a query sequence agains a database, or library of sequences. The database may be all the known sequences, stored in NCBI, a subset of these, or any other collection of sequences we want to search. The sequences in question may be nucleotide sequences, amino acids or even codons.
The result of a BLAST run is zero or more database sequences, which are similar enough to the query sequence. These results are called hits.
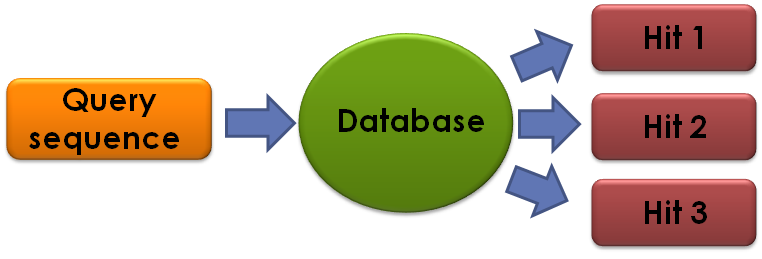
Lets run a nucleotide BLAST through the NCBI web, on the sequence:
GTTGAGATCACATAATAATTGATCGAGTTAATCTGGAGGATCAGTTTACTTTGGTCACCCATGGGCATTTGCT
ATTGCAGTGACCGAGATTTGCCATCGAGCCTCCTTGGGAGCTTTCTTGCTGGCGATCTAAACCCTAGCCCGGC
GCAGTTTTGCGCCAAGTCATATGACACATAATTGGCGAAGGGGGCGGCATGGTGCCTTGACCCTCCCCAAATC
ATTTTTTTAACAACTCTCAGCAACGGAAGGGCACGCCTGCCTGGGCATTGCGAGTCATATCTCTCCCTTAATG
AGGCTGTCCACACATACT
HTML('<iframe src=http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome width=1000 height=350></iframe>')
Let's take a look at the results.
First, we have a graphical representation of the best matches found in the database. Colors indicate the level of similarity.
Note that hits may contain more than one region of similarity. Each such region is called a high-scoring segment pair (HSP).
Then, a list of the hits follows. Each BLAST hit has its name, and several values describing the match:
- Max score - the score of the best HSP
- Total score - sum of scores for all HSPs (will be equal to Max score if the hit is only one HSP)
- Query cover - the percent of the query sequence covered by the hit
- E-value - the number of hits with better score expected to be found in a randomized database. This is used as an indication of the statistical significance of the match. The lower the E-value is, the more significant the hit. E-value < 10-4 are usually considered significant.
- Ident - percent of sequence identity between the hit and the query
Running BLAST from within python¶
We can perform the same BLAST run using a dedicated module included in Biopython. This is done using the qblast() method from the NCBIWWW module. It takes three arguments:
- BLAST algorithm to use (blastn,blastp,blastx, etc.), a string
- Database to querry (all nucleotides, refseq, all protein, etc.), a string
- Sequence - may be a string representing a sequence, a sequence object, a sequence-record object, a string representing sequence ID, or even a FASTA file.
from Bio.Blast import NCBIWWW
seq = Seq("GTTGAGATCACATAATAATTGATCGAGTTAATCTGGAGGATCAGTTTACTTTGGTCACCCATGGGCATTTGCTATTGCAGTGACCGAGATTTGCCATCGAGCCTCCTTGGGAGCTTTCTTGCTGGCGATCTAAACCCTAGCCCGGCGCAGTTTTGCGCCAAGTCATATGACACATAATTGGCGAAGGGGGCGGCATGGTGCCTTGACCCTCCCCAAATCATTTTTTTAACAACTCTCAGCAACGGAAGGGCACGCCTGCCTGGGCATTGCGAGTCATATCTCTCCCTTAATGAGGCTGTCCACACATACT")
try:
result_handle = NCBIWWW.qblast("blastn", "nt", seq)
print(result_handle)
except:
print('BLAST run failed!')
Parsing BLAST results¶
Once we have fetched the BLAST result, we would like to extract the information stored within it.
The first thing we do is to convert it into a usable format, using NCBIXML.read() on the result we retrieved.
from Bio.Blast import NCBIXML
blast_record = NCBIXML.read(result_handle)
print(blast_record)
still not very helpful. We will have to iterate over the BLAST hits, using .alignments:
for hit in blast_record.alignments:
print(hit)
within each hit, we can iterate over the HSPs, using .hsps:
for hit in blast_record.alignments:
print('***********************')
print(hit)
for hsp in hit.hsps:
print(hsp)
HSPs contain lots of useful information, for example, its name, length and E-value.
Let's write a function that will print only matches with E-value smaller than a chosen threshold.
def significant_matches(query,e_value_thresh):
try:
result_handle = NCBIWWW.qblast("blastn", "nt", query)
except:
print('BLAST run failed!')
return None
blast_record = NCBIXML.read(result_handle)
for hit in blast_record.alignments:
for hsp in hit.hsps:
if hsp.expect < e_value_thresh:
print(hit.title)
print(hsp.sbjct)
significant_matches(seq,e**(-100))
Class exercise 6C¶
Write a function that receives a sequences, runs a standard BLAST search and returns the name of the longest hit (use the .length and .title commands)
from Bio.Seq import Seq
from Bio.Blast import NCBIWWW, NCBIXML
def longest_blast_hit(seq):
result_handle = NCBIWWW.qblast("blastn", "nt", seq)
blast_record = NCBIXML.read(result_handle)
longest = 0
for hit in blast_record.alignments:
if hit.length > longest:
longest = hit.length
name = hit.title
return name
assert longest_blast_hit(seq).split('|')[1] == '47776119'
Fin¶
This notebook is part of the Python Programming for Life Sciences Graduate Students course given in Tel-Aviv University, Spring 2015.
The notebook was written using Python 3.4.1 and IPython 2.1.0 (download from PyZo).
The code is available at https://github.com//Py4Life/TAU2015/blob/master/lecture6.ipynb.
The notebook can be viewed online at http://nbviewer.ipython.org/github//Py4Life/TAU2015/blob/master/lecture6.ipynb.
This work is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License.
![]()