Previously on Py4Life¶
- Lists
- Dictionaries
- Functions
In today's episode¶
- Modules
- Files I/O
- The CSV format
- File parsing
- Regular expression
Modules¶
Last time, we learned how to write and use functions.
Luckily, we don't have to create all the code alone. Many people have written functions that perform various tasks.
These functions are grouped into packages, called modules, which are usually designed to address a specific set of tasks.
These functions are not always 'ready to use' by our code, but rather have to be imported into it.
Let's start with an example. Suppose we want to do some trigonometry, we can (in principal) write our own sin,cos,tan etc.. But it would be much easier to use the built-in math module.
# first, we import the module
import math
Now we can use values and functions within the math module
twice_pi = 2*math.pi
print(twice_pi)
radius = 2.3
perimeter = twice_pi * radius
print(perimeter)
print(math.sin(math.pi/6))
print(math.cos(math.pi/3))
If you only need one or two functions from a module, you can import them, instead of the whole module. This way, we don't have to call the module name every time.
# import required functions from math
from math import pi,sin,cos
# now we can use values and functions within the math module
twice_pi = 2*pi
print(twice_pi)
print(sin(pi/6))
print(cos(pi/3))
OK, cool, but how do I know which modules I need?

You can also view the module documentation to see what functions are available and how to use them. Each python module has a documentation page, for example: https://docs.python.org/3/library/math.html
Two more useful links:
https://pypi.python.org/pypi - a list of all Python modules
https://wiki.python.org/moin/NumericAndScientific - a list of scientific modules
Installing modules¶
Many Python modules are included within the core distribution, and all you have to do is import them. However, many other modules need to be downloaded and installed first.
Python has built-in tools for installing modules, but sometimes things go wrong. Therefore, try the following methods, in this order.
1. Use PIP¶
PIP is a built-in program which (usually) makes it easy to install packages and modules.
Since we can't access PIP from within a notebook, we'll use the Pyzo IEP shell, usually located at C:\pyzo\IEP.exe. This interactive shell can run usefull commands. It looks something like this:
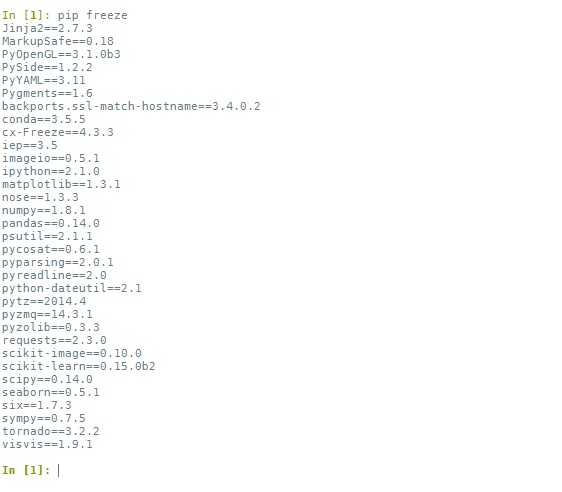
We'll enter our commands in this shell window. For example, to get a list of all the modules already installed (not including built-in modules) and their versions, we can type: pip freeze:
To install a new package, all we have to do is: pip install packagename, and that's it. Just make sure there are no error messages raised during the installation.
If, for some reason, things don't work out that well, proceed to option 2.
2. Use Conda¶
Conda is another useful tool, rather similar to PIP. To use it, just type conda in the IEP shell. To get the list of installed modules, type conda list. To install a module, type conda install packagename.
If this too doesn't work, proceed to option 3.
3. Use Windows binaries installation¶
If nothing else works, you can try looking for your package in this website. It contains many downloadable installers which you can just click through to easily install a package. Make sure to choose the download that fits your python version and operating system.
Not all modules are available through this website. If you don't find your module here, you might have to install from source. Details here
Files I/O¶
So far, we only used rather small data, like numbers, short strings and short lists. We stored these data in a local variable (i.e. in memory), and manipulated it. But what happens if we need to store large amounts of data?
- Whole genomes
- List of all insect species
- Multiple numeric values
This is what files are for!
Why do we need files?¶
- Store large amounts of data
- Use data in multiple sessions
- Use data outside python
- Provide data for other tools/programs
We'll start with simple text files and proceed to more complex formats.
Let's read the list of crop plants located in lec4_files/crops.txt
Reading files¶
Whenever we want to work with a file, we first need to open it. This is, not surprisingly, done using the open function.
This function returns a file object which we can then use.
crops_file = open('lec4_files/crops.txt','r')
The open function receives two parameters: the path to the file you want to open and the mode of opening (both strings). In this case - 'r' for 'read'.
Notice the / instead of \ in the path. This is the easiest way to avoid path errors. Also note that this command alone does nothing, just creates the file object (sometimes called file handle).
In fact, we'll usually use the open function differently:
with open('lec4_files/crops.txt','r') as crops_file:
# indented block
# do stuff with file
pass
OK, so what can we do with files?
The most common task would be to read the file line by line.
Looping over the file object¶
We can simply use a for loop to go over all lines. This is the best practice, and also very simple to use:
with open('lec4_files/crops.txt','r') as crops_file:
for line in crops_file:
if line.startswith('Musa'): # check if line starts with a given string
print(line)
Oops, why did we get double newlines?
Each line in the file ends with a newline character. Although it is invisible in most editors, it is certainly there! In python, a newline is represented as \n.
The print() command adds a new line to the newline character in the end of every line in the file, so we end up with double newlines.
We can use strip() to remove the character from the end of lines.
with open('lec4_files/crops.txt','r') as crops_file:
for line in crops_file:
line = line.strip()
if line.startswith('Musa'):
print(line)

Reading the entire file - read()¶
Another option is to read the entire file as a big string with the read() method.
Careful with this one! This is not recommended for large files.
with open('lec4_files/crops.txt','r') as crops_file:
entire_file = crops_file.read()
print(entire_file[:102]) # print first 102 characters
Reading line by line with readline()¶
The readline() method allows us to read a single line each time. It works very well when combined with a while loop, giving us good control of the program flow.
with open('lec4_files/crops.txt','r') as crops_file:
line = crops_file.readline() # read first line
while line:
line = line.strip()
if line.startswith('Triticum'):
print(line)
line = crops_file.readline() # read next line
REMEMBER to always read the next line within the while loop. Otherwise, you'll get stuck in an infinite loop, processing the first line over and over again...
There are other methods you can use to read files. For example, take a look at the readlines() method here:
https://docs.python.org/3/tutorial/inputoutput.html
Summary¶
Whenever treating a file, there are three elements:
- File path - the actual location of the file on the hard drive (use
/rather than\). - File object - the way files are handled in Python.
- File contents - what is extracted from the file, depending on the method used on the file object.
Class exercise 4A¶
Use one of the file-reading techniques shown above to:
- Print the last line in the file.
- Find out how many Garcinia species are in the file (use the
startswith()method).
with open('lec4_files/crops.txt','r') as crops_file:
entire_file = crops_file.read()
lines_list = entire_file.split('\n')
print(lines_list[-1])
with open('lec4_files/crops.txt','r') as crops_file:
triticum_count = 0
for line in crops_file:
if line.startswith('Garcinia'):
triticum_count += 1
print(triticum_count)
Writing to a file¶
To write to a file, we first have to open it for writing. This is done using one of two modes: 'w' or 'a'.
'w', for write, will let you write into the file. If it doesn't exist, it'll be automatically created. If it exists and already has some content, the content will be overwritten!
'a', for append, is very similar, only it will not overwrite, but add your text to the end of an existing file.
with open('lec4_files/output.txt','w') as out_file:
# indented block
# write into file...
pass
Writing is done using good, old print(), only we add the argument file = <file object>.
with open('lec4_files/output.txt','w') as out_file:
print('This is the first line', file=out_file)
line = 'Another line'
print(line, file=out_file)
seq1 = 'ATTAGCGGATA'
seq2 = 'GGCATATAT'
print(seq1 + seq2, file=out_file)
Parsing files¶
Parsing is "the process of analyzing a string of symbols, either in natural language or in computer languages, conforming to the rules of a formal grammar." (definition from Wikipedia).
More simply, parsing is reading a file in a specific format, 'slurping' the data and storing it in a data structure of your choice (list, dictionary etc.). We can then use this structure to analyze, print or simply view the data in a certain way.
Each file format has its own set of 'rules', and therefore needs to be parsed in a tailored manner. Here we will see an example very relevant for biologists.
The FASTA format¶
FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences, in which nucleotides or amino acids are represented using single-letter codes. Each sequence has a header. Header lines start with '>'.
The file camelus.fasta includes five sequences of Camelus species. In this parsing example, we'll arrange the data in this file in a dictionary, so that the key is the id number from the header, and the value is the sequence.
from IPython.display import FileLink
FileLink('lec4_files/camelus.fasta')
We'll start by writing the parsing function.
def parse_fasta(file_name):
"""
Receives a path to a fasta file, and returns a dictionary where the keys
are the sequence IDs and the values are the sequences.
"""
# create an empty dictionary to store the sequences
sequences = {}
# open fasta file for reading
with open(file_name,'r') as f:
# Loop over file lines
for line in f:
# if header line
if line.startswith('>'):
seq_id = line.split('|')[1]
# if sequence line
else:
seq = line.strip()
sequences[seq_id] = seq
return sequences
Now we can use the result. For example, let's print the first 10 nucleotides of every sequence.
camelus_seq = parse_fasta('lec4_files/camelus.fasta')
for seq_id in camelus_seq:
print(seq_id," - ",camelus_seq[seq_id][:10])

Class exercise 4B¶
Change the function above so that it takes the gb accession (e.g. EF471324.1) as key and 30 first nucleotides as value. Then use the output dictionary to print the results to a new file, in the following format:
EF471324.1: AGAGTCTTTGTAGTATATGGATTACGCTGG
EF471323.1: AGAGTCTTTGTAGTATATTGATTACGCTGG
.
.
.
# new function
def parse_fasta_30_nuc(file_name):
"""
Receives a path to a fasta file, and returns a dictionary where the keys
are the sequence gb accession numbers and the values are the first 30
nucleotides of the sequences.
"""
# create an empty dictionary to store the sequences
sequences = {}
# open fasta file for reading
with open(file_name,'r') as f:
# Loop over file lines
for line in f:
# if header line
if line.startswith('>'):
gb = line.split('|')[3]
# if sequence line
else:
seq = line.strip()[:30]
sequences[gb] = seq
return sequences
# parse file
camelus_seq = parse_fasta_30_nuc('lec4_files/camelus.fasta')
# write to new file
with open('lec4_files/4b_output.txt','w') as of:
for gb_id in camelus_seq:
print(gb_id + ':',camelus_seq[gb_id], file=of)
Regular expressions¶
Parsing files can sometimes be done using only the string class methods, as we did above. However, sometimes it can get tricky. Let's take an example.
DNA patterns¶
Suppose we have a DNA sequence in which we want to look for a specific pattern, say, 'TATAGGA'.
What do we do?
Easy, we use the find method.
seq = "ccgcaattcactctataggagcaggaacatggataaagctcacagtcgca"
if seq.find('tatagga') >= 0:
print('pattern found!')
OK, but what if we need to look for a more flexible pattern, such as 'TATAGGN'?
We can do:
if seq.find('tatagga') >= 0 or seq.find('tataggt') >= 0 or seq.find('tataggc') >= 0 or seq.find('tataggg') >= 0:
print('pattern found!')
But that's lots of work and also, what if we need 'TATAGNN'?
There are too many combinations to cover manually!
What we need is a more general way of doing such matching. This is what Regular expressions are for!
What are regular expressions?¶
Regular expressions (regex) are sets of characters that represents a search pattern. It's like a specific language that was designed to tell us how a text string should look. It includes special symbols which allow us to depict flexible strings.
This is a very powerful tool when looking for patterns or parsing text.
We'll soon see what we can do with it and how to use it.
Using regular expressions¶
In order to use regex, we need to use pythons built-in dedicated module. That means we don't have to install anything, just import the re module.
import re
Raw strings¶
We've already encountered some special characters, such as \n (newline) and \t (tab).
In regular expressions we want to avoid any confussion, and therefore use a special notation, telling python that we have no buisness with special characters here. We simply put an r outside the quotation marks.
normal_string = "There will be\na new line"
raw_string = r"There won't be\na new line"
print(normal_string)
print(raw_string)
ALWAYS use raw strings when working with regular expressions!
Searching for patterns¶
This is the most basic task regex is used for. We just want to know if a pattern can be found within a string.
The first step when working with regex is always compiling. This means we transform a simple string such as 'tatagga' into a regex pattern. This is done using re.compile()
regex = re.compile(r'tatagga') # notice the 'r'
We didn't match anything yet, just prepared the regex pattern. Once we have it, we can use it to seqrch within another string. For this we can use the re.search() method. It takes two parameters: regex and string to search ('target string') and returns True if the pattern was found and False otherwise.
if re.search(regex,seq):
print('pattern found!')
Character groups¶
The last example wasn't particularly useful, right?
OK, so here's when it gets interesting. We can define character groups within our regex, so that any of them will be matched. We do that using square brackets, and put all possible matches within them. So if we want to match 'TATAGGN' we'll do:
regex = re.compile(r'tatagg[atgc]')
if re.search(regex,seq):
print('pattern found!')
if re.search(regex,"tataggn"):
print('pattern found!')
We can put any list of characters within the brackets. There are also a few tricks to make things easier:
- [0-9] - any digit
- [a-z] - any letter
- [a-p] - any letter between a and p
There are also special symbols for common groups:
- \d - any digit (equivalent to [0-9])
- \w - any 'word' character - letters, digits and underscore (equivalent to [a-zA-Z0-9_)
- \s - any whitespace character - space, tab, newline and other weird stuff (equivalent to [ \t\n\r\f\v])
And finally, there's the wildcard symbol, represented by a dot (.).
This means any character (except for a newline).
Careful with this one! It'll take almost anything, so use it wisely.
# examples:
regex = re.compile(r'\d[d-k][2-8].')
if re.search(regex,'7f6,'):
print('pattern found!')
if re.search(regex,'hello7f6world'):
print('pattern found!')
if re.search(regex,'5l7o'):
print('pattern found!')
if re.search(regex,'7f6'):
print('pattern found!')
Being negative¶
Sometimes we want to tell python to search for 'anything but...'. We can do that in two ways:
If we are using character groups in square brackets, we can simply add a cadet (^) before the characters. For example [^gnp%] means 'match anything but 'g','n','p' or '%''. If we are using the special character groups, we can replace the symbol with a capital letter, so for example \D means 'match anything but a digit'.
regex = re.compile(r'AAT[^G]TAA')
if re.search(regex,'AATCTAA'):
print('pattern found!')
if re.search(regex,'AATGTAA'):
print('pattern found!')
regex = re.compile(r'AAT\STAA')
if re.search(regex,'AATCTAA'):
print('pattern found!')
if re.search(regex,'AATGTAA'):
print('pattern found!')
if re.search(regex,'AAT TAA'):
print('pattern found!')
Alteration¶
When we want to create multiple options for longer patterns, character groups are not enough. In these cases we have to use the special '|' (pipe) character, which simply means 'or'.
For example, if we want to match a pattern that starts with AGG, then either CCG or TAG, and finally GTG, we can do:
regex = re.compile(r'AGG(CCG|TAG)GTG')
if re.search(regex,'AGGTAGGTG'):
print('pattern found!')
if re.search(regex,'AGGCCGGTG'):
print('pattern found!')
if re.search(regex,'AGGCCTGTG'):
print('pattern found!')
Repetition¶
In many cases, we want to write regular expressions where a part of the pattern repeats itself multiple times. For that, we use quantifiers.
If we know exactly how many repetitions we want, we can use {<number>}:
regex = re.compile(r'GA{5}T')
if re.search(regex,'GAAAAAT'):
print('pattern found!')
if re.search(regex,'GAAAT'):
print('pattern found!')
We can also set an acceptable range of number of repeats, which is done using {<minimum repeats>,<maximum repeats>:
regex = re.compile(r'GA{3,5}T')
if re.search(regex,'GAAAAAT'):
print('pattern found!')
if re.search(regex,'GAAAT'):
print('pattern found!')
if re.search(regex,'GAAAAAAT'):
print('pattern found!')
To say 'x or more repetitions', we use {x,}. For 'up to x repetitions', we can use {0,x}.
For more general cases, there are three special symbols we can use:
- + - repeat 1 or more times
- * - repeat 0 or more times
- ? - repeat 0 or 1 times, or in other words 'optional' character.
regex = re.compile(r'GA+TT?[AC]*')
if re.search(regex,'GAATTACCA'):
print('pattern found!')
if re.search(regex,'GATACCA'):
print('pattern found!')
if re.search(regex,'GTACCA'):
print('pattern found!')
if re.search(regex,'GAAAAAAAT'):
print('pattern found!')
Note 1: Quantifiers always refer to the character that appears right before them. This could be a normal character or a character group. If we want to indicate a repeat of several characters, we enclose them in ().
regex = re.compile(r'GGCG(AT)+GGG')
if re.search(regex,'GGCGATATATATGGG'):
print('pattern found!')
if re.search(regex,'GGCGATTAATGGG'):
print('pattern found!')
regex = re.compile(r'GGCG(AT)?GGG')
if re.search(regex,'GGCGATGGG'):
print('pattern found!')
if re.search(regex,'GGCGGGG'):
print('pattern found!')
Note 2: Whenever we want to match one of the special regex characters in its 'normal' context, we simply put a '' before it. For example: \*, \+, \{...
regex = re.compile(r'.+\{\d+\}\.')
sentence = 'A sentence that ends with number in curly brackets {345}.'
if re.search(regex,sentence):
print('pattern found!')
Class exercise 4D¶
The code below includes a list of made-up gene names. Complete it to only print gene names that satisfy the following criteria:
- Contain the letter 'd' or 'e'
- Contain the letter 'd' and 'e', in that order (not necessarily in a row)
- Contain three or more digits in a row
import re
genes = ['xkn59438', 'yhdck2', 'eihd39d9', 'chdsye847', 'hedle3455', 'xjhd53e', '45da', 'de37dp','map492ty']
# 1.
print('Gene names containing d or e:')
regex1 = re.compile(r'[de]')
for gene in genes:
if re.search(regex1,gene):
print(gene)
print('------------------------')
# 2.
print('Gene names containing d and e, in that order:')
regex2 = re.compile(r'd[^e]*e')
for gene in genes:
if re.search(regex2,gene):
print(gene)
print('------------------------')
# 3.
print('Gene names containing three digits in a row:')
regex3 = re.compile(r'\d{3,}')
for gene in genes:
if re.search(regex3,gene):
print(gene)
Enforcing positions¶
We can enforce the a regex to match only the start or end of the input string. We do that by using the ^ and $ symbols, respectively.
regex = re.compile(r'^my name')
if re.search(regex,'my name is Slim Shady'):
print('pattern found!')
if re.search(regex,'This is my name'):
print('pattern found!')
regex = re.compile(r'my name$')
if re.search(regex,'This is my name'):
print('pattern found!')
We can combine the start and end symbols to match a whole string:
regex = re.compile(r'^GC[GTC]{2,10}TTA$')
if re.search(regex,'GCTTCGCTTA'):
print('pattern found!')
if re.search(regex,'GCTTCGCTTAG'):
print('pattern found!')
Extracting matches¶
OK, now that we know the 'language' of regular expression, let's see another useful thing we can do with it.
So far, we only used regex to test if a string matches a pattern, but sometimes we also want to extract parts of the string for later use.
Let's take an example.
The GATA-4 Transcription factor¶
GATA-4 is a TF in humans, known to have an important role in cardiac development (Oka, T., Maillet, M., Watt, A. J., Schwartz, R. J., Aronow, B. J., Duncan, S. A., & Molkentin, J. D. (2006). Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circulation research, 98(6), 837-845.)
It is also known to bind the motif: AGATADMAGRSA (where M = A or C, D = A,G or T, R = A or G and S = C or G).
Using regex, it's easy to write a function that checks if a sequence includes this motif.

def check_for_GATA4(sequence):
motif_regex = re.compile(r'AGATA[AG][AC]AG[AG][CG]A')
if re.search(motif_regex,sequence):
return True
else:
return False
test_seq1 = 'AGAGTCTTTGAGATAGCAGACATAGTATATGGATTACGCTGGTCTTGTAAACCATAAAAGGAGAGCCACACTCTCCCTAAGACTCAGGGAAGAGGCCAAAGCCCCACCACCAGCACCCAAAGCTG'
check_for_GATA4(test_seq1)
test_seq2 = 'AGAGTCTTTGAGATAGTAGACATAGTATATGGATTACGCTGGTCTTGTAAACCATAAAAGGAGAGCCACACTCTCCCTAAGACTCAGGGAAGAGGCCAAAGCCCCACCACCAGCACCCAAAGCTG'
check_for_GATA4(test_seq2)
But what if we want to extract the actual sequence that matches the regex?
Let's have another look at the re.search() method. So far, we only used it to test if a match exists or not. But it actually returns something, which we can use to get the exact match, with the group() method.
This method is used on the search result to get the match. So the following function will return the actual match in the sequence, if one exists. Otherwise, it will return None.
def find_GATA4_motif(sequence):
motif_regex = re.compile(r'AGATA[AG][AC]AG[AG][CG]A')
result = re.search(motif_regex,sequence) # notice the assignment here
if result is None:
return None
else:
return result.group()
print(find_GATA4_motif(test_seq1))
print(find_GATA4_motif(test_seq2))
Since most of the motif is fixed, we might only be interested in the 'ambiguous' parts (that is, the DM part and the RS part). We can capture specific parts of the pattern by enclosing them with parentheses. Then we can extract them by giving the group() method an argument, where '1' means 'extract the first captured part', '2' means 'extract the second captured part' and so on. The following function will capture the ambiguous positions and return them as elements of a list.
def extract_ambiguous_for_GATA4(sequence):
motif_regex = re.compile(r'AGATA([AG])([AC])AG([AG])([CG])A') # notice the parentheses
result = re.search(motif_regex,sequence)
if result is None:
return None
else:
D = result.group(1)
M = result.group(2)
R = result.group(3)
S = result.group(4)
return [D,M,R,S]
D,M,R,S = extract_ambiguous_for_GATA4(test_seq1)
print('D nucleotide:',D)
print('M nucleotide:',M)
print('R nucleotide:',R)
print('S nucleotide:',S)
More on regular expression¶
There are some other cool things we can do with regex, which we'll not discuss here:
- Split strings by regex
- Substitute parts of string using regex
- Get the position in the string where a pattern was found
If you want to do any of these, take a look at the re module documentation
https://docs.python.org/3/library/re.html
Class exercise 4E¶
The 'GATA4_promoters.fasta' file includes (made-up) promoter sequences for genes suspected to be regulated by GATA-4.
We'll use everything we've learned so far to write a program that summarizes some interesting statistics regarding the GATA-4 motifs in these promoters.
First, let's adjust the parse_fasta() function we created earlier for the specif format of the promoters file:
def parse_promoters_fasta(file_name):
"""
Receives a path to a fasta file, and returns a dictionary where the keys
are the sequence names and the values are the sequences.
"""
# create an empty dictionary to store the sequences
sequences = {}
# open fasta file for reading
with open(file_name,'r') as f:
# Loop over file lines
for line in f:
# if header line
if line.startswith('>'):
seq_id = line[1:-1] # take the whole line, except the '>' in the beginning and '\n' at the end
# if sequence line
else:
seq = line.strip()
sequences[seq_id] = seq
return sequences
Write a function that receives a promoters fasta dictionary, and counts how many of the promoters have the GATA-4 motif. Use any of the functions defined above and complete the code:
def count_promoters_with_motif(promoters_dictionary):
"""
Receives a dictionary representing a promoters fasta file,
and counts how many of the promoters include a GATA-4 motif.
"""
promoters_count = 0 # store the number of promoters with GATA-4 motif
for p in promoters_dictionary:
if check_for_GATA4(promoters_dictionary[p]):
promoters_count += 1
return promoters_count
- For promoters that do include the GATA-4 motif, we would like to know the frequencies of the different nucleotides for each of the four variable positions in the motif. Complete the code:
def get_positions_statistics(promoters_dictionary):
"""
Receives a dictionary representing a promoters fasta file,
and returns the frequencies of possible nucleotides in
each variable position.
"""
# define a dictionary for each position, to store the nucleotide frequencies
# D position
D_dict = {'A':0, 'G':0, 'T':0}
# M position
M_dict = {'A':0, 'C':0}
# R position
R_dict = {'A':0, 'G':0}
# S position
S_dict = {'C':0, 'G':0}
# itterate over promoters
for p in promoters_dictionary:
# if promoter includes the GATA-4 motif
if check_for_GATA4(promoters_dictionary[p]):
# get variable nucleotides in promoter
D,M,R,S = extract_ambiguous_for_GATA4(promoters_dictionary[p])
# insert to dictionaries
D_dict[D] += 1
M_dict[M] += 1
R_dict[R] += 1
S_dict[S] += 1
return D_dict, M_dict, R_dict, S_dict
- Now, we just have to write a function that will summarize the results in a CSV file. It should receive the frequencies dictionaries and write statistics to an output file. Complete the code:
def summarize_results(D_dict, M_dict, R_dict, S_dict, output_file):
with open(output_file, 'w') as fo:
csv_writer = csv.writer(fo)
# write headers line
csv_writer.writerow(['Position','A','G','C','T'])
# summarize D position
csv_writer.writerow(['D',D_dict['A'],D_dict['G'],0,D_dict['T']])
# summarize M position
csv_writer.writerow(['M',M_dict['A'],0,M_dict['C'],0])
# summarize R position
csv_writer.writerow(['R',R_dict['A'],R_dict['G'],0,0])
# summarize S position
csv_writer.writerow(['S',0,S_dict['G'],S_dict['C'],0])
- Now that we have all the functions ready, we can write the main program. Complete the code:
import csv
promoters_file = "lec4_files/GATA4_promoters.fasta"
output_file = "lec4_files/promoters_stats.csv"
# parse fasta file
promoters_dict = parse_promoters_fasta(promoters_file)
# Count promoters with/without GATA-4 motif
promoters_with_motif = count_promoters_with_motif(promoters_dict)
promoters_without_motif = len(promoters_dict) - promoters_with_motif
print('Total promoters:',promoters_with_motif + promoters_without_motif)
print('Promoters with GATA-4 motif:',promoters_with_motif)
print('Promoters without GATA-4 motif:',promoters_without_motif)
# Get statistics
D_dict, M_dict, R_dict, S_dict = get_positions_statistics(promoters_dict)
# write to CSV
summarize_results(D_dict, M_dict, R_dict, S_dict,output_file)
The CSV format¶
Comma separated values (CSV) is a very common and useful format for storing tabular data. It is similar to an Excel file, only it is completely text based. Let's have a look at an example file, both using Excel and a simple text editor.
We can, quite easily, create our own functions for dealing with CSV files, for example by splitting each line by commas. However, Python has a built-in module for exactly this purpose, so why bother?
Reading CSV files¶
The most simple way to read a CSV file is to use the modules reader function. This function receives a file object (created with open()) and returns a reader object.
import csv
experiments_file = 'lec4_files/electrolyte_leakage.csv'
with open(experiments_file, 'r') as f:
csv_reader = csv.reader(f)
Once we have defined the csv reader, we can use it to iterate over the file lines. Each row is returned as a list of the column values.
experiments_file = 'lec4_files/electrolyte_leakage.csv'
with open(experiments_file, 'r') as f:
csv_reader = csv.reader(f)
for row in csv_reader:
print(row[0])
Writing CSV files¶
Writing is also rather straightforward. The csv module supplies the csv.writer object, which has the method writerow(). This function receives a list, and prints it as a csv line.
new_file = 'lec4_files/out_csv.csv'
with open(new_file, 'w', newline='') as fo: # notice the 'w' instead of 'r'
csv_writer = csv.writer(fo)
csv_writer.writerow(['these','are','the','column','headers'])
csv_writer.writerow(['and','these','are','the','values'])
Class exercise 4C¶
The electrolyte_leakage.csv file depicts the results of experiments on different Arabidopsis ecotypes (accessions). In each row, there are 3 control plants and 3 plants tested under draught stress.
Read the CSV file, calculate the mean result for control and for test plants of each ecotype, and print the result as a new CSV file, in the following way:
Accession | control mean | test mean
101AV/Ge-0 | 7.34 | 3.03
157AV/Ita-0| 16.85 | 2.92
.
.
.
Use the provided accessory function to calculate means.
Try opening the output file in Excel to make sure your code works propperly.
def mean_of_string_values(lst):
"""
receives a list of strings representing numbers and returns their mean
"""
numeric_lst = []
for x in lst:
numeric_lst.append(float(x))
return mean(numeric_lst)
experiments_file = 'lec4_files/electrolyte_leakage.csv'
with open(experiments_file, 'r') as f:
with open('lec4_files/4c_output.csv','w', newline='') as fo:
csv_writer = csv.writer(fo)
csv_writer.writerow(['Accession','control mean','test mean'])
csv_reader = csv.reader(f)
next(csv_reader)
for row in csv_reader:
acc = row[0]
control = row [1:4]
test = row[4:]
to_write = [acc,mean_of_string_values(control),mean_of_string_values(test)]
csv_writer.writerow(to_write)
Fin¶
This notebook is part of the Python Programming for Life Sciences Graduate Students course given in Tel-Aviv University, Spring 2015.
The notebook was written using Python 3.4.1 and IPython 2.1.0 (download from PyZo).
The code is available at https://github.com//Py4Life/TAU2015/blob/master/lecture4.ipynb.
The notebook can be viewed online at http://nbviewer.ipython.org/github//Py4Life/TAU2015/blob/master/lecture4.ipynb.
The notebook is also available as a PDF at https://github.com//Py4Life/TAU2015/blob/master/lecture4.pdf?raw=true.
This work is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License.
![]()