Merged PTM and Expression SCLC Cluster¶
This notebook will investigate the cluster of up-regulated PTMs and genes in SCLC lung cancer cell lines. This cluster was isolated and saved in the notebook: CST_Data_Viz.ipynb.
from clustergrammer_widget import *
net = Network(clustergrammer_widget)
# load data
net.load_file('histology_clusters/merge_sclc.txt')
merge_sclc = net.export_df()
# manually set category colors for rows and columns
net.set_cat_color('row', 1, 'Data-Type: phospho', 'red')
net.set_cat_color('row', 1, 'Data-Type: Rme1', 'purple')
net.set_cat_color('row', 1, 'Data-Type: AcK', 'blue')
net.set_cat_color('row', 1, 'Data-Type: Kme1', 'grey')
net.set_cat_color('row', 1, 'Data-Type: Exp', 'yellow')
net.set_cat_color('col', 1, 'Histology: SCLC', 'red')
net.set_cat_color('col', 1, 'Histology: NSCLC', 'blue')
net.set_cat_color('col', 2, 'Sub-Histology: SCLC', 'red')
net.set_cat_color('col', 2, 'Sub-Histology: NSCLC', 'blue')
net.set_cat_color('col', 2, 'Sub-Histology: squamous_cell_carcinoma', 'yellow')
net.set_cat_color('col', 2, 'Sub-Histology: bronchioloalveolar_adenocarcinoma', 'orange')
net.set_cat_color('col', 2, 'Sub-Histology: adenocarcinoma', 'grey')
Cluster of Up-regulated PTMs and Genes in SCLC Cell Lines¶
Below we will visualize the SCLC cluster. We will highlight a few of the interesting genes/proteins and sub-clusters found in this cluster of up-regulated PTMs and gene expression levels data in SCLC cell lines.
net.cluster(views=[])
net.widget()
We can see that the cluster is composed of roughly for smaller clusters that are generally up-reglated in SCLC cell lines and down-regulated in NSCLC cell lines. We can also see that the cluster is composed of a mixture of phosphorylation, expression, methylation and acetylation.
NKX2-1 and SOX2 Cluster¶
NKX2-1 is a transcription factor that is known to have a role in lung development and has been used as a biomarker in lung cancer (Yang et al. 2012). In this cluster we find NKX2-1 expression and NKX2-1 methylation (R121) are both present and cluster closely together. The expression and methylation data are from independent sources and this NKX2-1 co-clustering demonstrate broad agreement between the two datasets. The data imply that variability in NKX2-1 methylation may be the result of variability in its expression level. Below is a screenshot of the immediate cluster of phophorylation, expression, and methylation including NKX2-1:
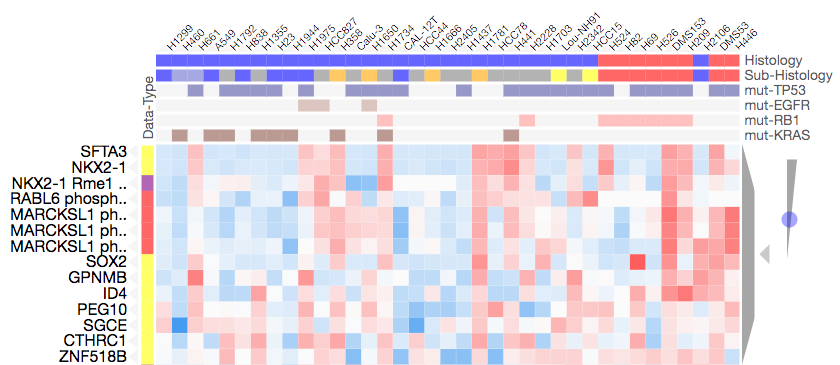
NKX2-1 levels are known to be generally high in SCLC cell lines which broadly agrees with our results. NKX2-1 also clusters with several other lung associated genes including:
- STFA3 is a lung associated protein with possible immune functions (Schict et al. 2014)
- SOX2 is known to be involved in lung cancer (Karachaliou et al. 2013) and is also thouhgt to interact with NKX2-1 (Ferri et al. 2013)
- GPNMB is thought to be involved in lung cancer (Oyewumi et al. 2016)
- ID4 has been found to be associated with lung cancer metastisis (Pan et al. 2015)
- PEG10 is thought to play a role in lung cancer proliferation (Deng et al 2014)
- CTHRC1 overexpression is though to be associated with NSCLC tumor aggressiveness (Ke et at. 2014).
This cluster also includes MARCKSL1 (MARCKS-like 1) phosphorylations: S71, T148, and S151. MARCKSL1 is thought to play a role in cytoskeletal reglation see Gene Cards. This implies that phosphorylation of MARCKSL1 phosphorylation may play a role in lung development based on its co-clustering with NKX2-1 expression/methylation.
Cytoskeletal and Extracellular Matrix Proteins¶
Several cytoskeletal and extracellular matrix (ECM) associated genes/proteins appear in this cluster. This highlights the importance cytoskeletal/ECM function in SCLC lung cancer.
- Actin: ACTB is acetylated at K328, K61.
- Tubulin: TUBA1A exp, TUBA1A Ack K394, TUBB phos S115,
- Collagens: COL11A1, COL1A2, COL4A1, COL6A3 (expression and correlation)
- Rac GTPase: RACGAP1 (Rac GTPase activiting protein 1) is phosphorylated at T606 (regulates actin)
- Myosin: MYH9 acetylated, MYH10 acetylated, MYH11 acetylated, MYLK phosphorylated, MYO9A phos
- Stathmin a microtubule depolymerizer: STMN1 Ack K53, STMN2-exp (stathmin). Stathmin expression is thought to be a poor prognostic biomarker in NSCLC (Nie et al. 2015). Stathmin 2 expression and Stathmin 1 acetylation are similar.
- Microtubule associated proteins: MAP2 exp: thought to be involved in MT assembly and neurogenesis. MAP7, primarily expressed in epithelial. DCX, doublecortin, microtubule binding and involved in the development of the cortex.
Gene Ontology Biological Process 2015¶
Here we are pre-calculating enrichment for biological processes from the Gene Ontology resource using the enrichrgram method (see Clustergrammer-PY's API for more information). This will help us understand the broad biological processes occurring in this cluster of up-regulated PTMs and Genes.
net.enrichrgram('GO_Biological_Process_2015')
net.cluster(views=[])
net.widget()
mRNA Processing and Gene Expression¶
We see enrichment for mRNA processing, splicing, and gene expression. We also see similar results with KEGG 2016 enrichment, which can be run in this notebook with the Enrichrgram button.
Neuronal Sub-Cluster¶
Genes with the above ontologies are mainly distributed at the top and bottom sub-clusters - note that the largest middle sub-cluster with highly up-regulated values has relatively few genes with this association. We can investigate the biological processes in the middle sub-cluster by clicking the dedrogram crop button and re-running the enrihment analysis using the front-end Enrichrgram button.
Doing so reveals that this cluster is also enriched for gene expression and mRNA processing but also for several neuronal processes including: neuron projection, axon guidance and neuron projection morphology. This agrees with the neuronal characteristics of SCLC cell lines (see Onganar et al 2005).
Disease Perturbations from GEO Up¶
Using the 'Disease Perturbations from GEO Up' Enrichr library we can find diseases that have up-regulated genes that are similar to our up-regulated genes/proteins. This can help us understand the disease-associations of the differntially expressed genes and proteins with differentially reglated PTMs.
net.enrichrgram('Disease_Perturbations_from_GEO_up')
net.cluster(views=[])
net.widget()
Above, we see that up-regulated genes and PTMs in SCLC are similar to up-regulated genes in the following diseases: oligodendroglioma, multiple sclerosis, and other cancers.
MGI Mammalian Phenotype¶
To find phenotypes associated with our genes/proteins, we can use the 'MGI Phenotype Level 4' Enrichr library. This library will return phenotypes that are associated with gene knockout in mice. This can give us a less biased overview of gene/phenotype relationships.
net.enrichrgram('MGI_Mammalian_Phenotype_Level_4')
net.cluster(views=[])
net.widget()
Perinatal Lethality and Neuronal Abnormalities¶
Above we see that our genes/proteins are enriched for the following broad phenotypes:
- perinatal lethality (prenatal, preweaning, and postnatal lethality)
- neuronal abnormalities (neuron morphology, brain morphology, spinal cord morphology, and nervous system abnormalities).
The enrichment for neuronal abnormality phenotypes agrees with the neuronal enrichment we obtained above with Gene Ontology Biological Process and with our prior knowledge of SCLC characteristics.