Lung Cancer Post-Translational Modification and Gene Expression Regulation¶
Lung cancer is a complex disease that is known to be regulated at the post-translational modification level, e.g. phosphorylation driven by kinases. Our collaborators at Cell Signaling Technology Incorporated (CST) used Tandem Mass Tag (TMT) mass spectrometry to measure differential phosphorylation, acetylation, and methylation in a panel of 42 lung cancer cell lines compared to non-cancerous lung tissue. Gene expression data from 37 of these lung cancer cell lines was also independently obtained from the publically available Cancer Cell Line Encyclopedia (CCLE). In this notebook we will analyze PTM and gene expression regulation across these 37 common lung cancer cell lines. This notebook is part of the GitHub repo: MaayanLab/CST_Lung_Cancer_Viz. The post-translational modification (PTM) and gene expression data were pre-processed (normalized, filtered, etc) in the CST_Data_Processing.ipynb notebook.
In this notebook we will
- visualize PTM, Expression, and merged PTM-Expression datasets
- identify clusters of PTMs/genes
- use enrichment analysis to understand the biological processes involved in PTM/gene-expression clusters
Lung Cancer Histology¶
The lung cancer cell lines in this joint dataset fall into two histologies: Non-small Cell Lung Cancer (NSCLC), and Small Cell Lung Cancer (SCLC). NSCLC is more common than SCLC and refer to all epithelial lung cancers other than SCLC. SCLC is highly malignant and develops metastasis earlier than NSCLC. SCLC is thought to originate from neuroendocrine cells and are known to have neuronal characteristics Onganer et al. 2005.
Load Data and Clustergrammer-Widget¶
First, we will make an instance of the Clustergrammer-PY Network class that will be used to load, analyze, and visualize our data. For more information see Clustergrammer-PY API and Clustergrammer-Widget.
# make instance of Clustergrammer's Network object and pass in the clustergrammer_widget class
from clustergrammer_widget import *
net = Network(clustergrammer_widget)
# load our data
net.load_file('../lung_cellline_3_1_16/lung_cl_all_ptm/precalc_processed/CST_CCLE_ptm.txt')
# check shape of data
print('PTM data shape: '+ '' + str(net.dat['mat'].shape))
PTM data shape: (1730, 37)
CST Post Translational Modification Lung Cancer Data¶
Here we will visualize pre-processed PTM data from our collaborators at CST (see CST_Data_Processing.ipynb for more information on data pre-processing). In this dataset differential phosphorylation, methylation, and acetylation were measured relative to non-cancerous lung tissue across 37 lung cancer cell lines (note, not all PTM types had the same number of measurements, e.g. phosphorylation had the most measurements). PTM levels were quantile normalized in each cell, PTMs with more than 7 missing values were disguarded, and PTM levels across cell lines wre Z-score normalized to highlight differential regulation across lung cancer cell lines. Cell line 'meta-data' is shown as column categories and include: Histology, TP53 mutation, EGFR mutation, RB1 mutation, and KRAS mutation. Cell line metadata was obtained from our collaborators at CST and from metadata available from the CCLE.
# manually set category colors for rows and columns
net.set_cat_color('row', 1, 'Data-Type: phospho', 'red')
net.set_cat_color('row', 1, 'Data-Type: Rme1', 'purple')
net.set_cat_color('row', 1, 'Data-Type: AcK', 'blue')
net.set_cat_color('row', 1, 'Data-Type: Kme1', 'grey')
net.set_cat_color('col', 1, 'Histology: SCLC', 'red')
net.set_cat_color('col', 1, 'Histology: NSCLC', 'blue')
net.set_cat_color('col', 2, 'Sub-Histology: SCLC', 'red')
net.set_cat_color('col', 2, 'Sub-Histology: NSCLC', 'blue')
net.set_cat_color('col', 2, 'Sub-Histology: squamous_cell_carcinoma', 'yellow')
net.set_cat_color('col', 2, 'Sub-Histology: bronchioloalveolar_adenocarcinoma', 'orange')
net.set_cat_color('col', 2, 'Sub-Histology: adenocarcinoma', 'grey')
net.cluster(views=[])
net.widget()
Cell Lines Cluster According to Histology and Mutation¶
Above, we see that cell lines cluster according to their histology - almost all NSCLC cell lines (blue column category) cluster together (except for H2106) and almost all SCLC cell lines (red column category) cluster together. Cell lines also cluster to some extent based on their Sub-Histology (e.g. squamous cell carcinoma cells in yellow and bronchioloalveolar adenocarcinoma in orange) and based on mutation. We also see that cell lines appear to cluster according to common mutations in: EGFR, KRAS, and RB1. Mutations in these genes may be the drivers behind their common PTM regulation.
PTM Clusters¶
We can see two high-level clusters of PTMs that have either high/low levels in SCLC cell lines and low/high levels, respectively in NSCLC cell lines. The cluster with high levels in SCLC cell lines is mainly composed of phosphorylation, arginine methylation, and lysine acetylation, while the cluster with low levels in SCLC cell lines is almost entierly composed of phosphorylation (red row category). Below, we will use the interactive dendrogram in combination with the widget_df method (see Clustergrammer-PY API) to export these clusters to TSV for further analysis.
PTM cluster according to Data-Type¶
We see that PTMs (rows) also tend to cluster according to their type (e.g. phosphorylation). However, we also see that different types of modifications co-cluster.
# # here we are using the interactive dendrogram (not shown) to select clusters and export them to TSVs using
# # the widget_df method.
# ptm_sclc = net.widget_df()
# ptm_nsclc = net.widget_df()
# ptm_sclc.to_csv('histology_clusters/ptm_sclc.txt', sep='\t')
# ptm_nsclc.to_csv('histology_clusters/ptm_nsclc.txt', sep='\t')
Gene Expression Data¶
We obtained gene expression data from the Cancer Cell Line Encyclopedia (CCLE) for 37 lung cancer cell lines assayed by our collaborators at CST. This independent dataset can be used to find novel correlations between differentially expressed genes and PTMs as well as determine whether lung cancer cell lines behave similarly in gene-expression-space and PTM-space. The gene expression data was processed in the CST_Data_Processing.ipynb notebook that: kept the top 1000 genes with the greatest variance across the cell lines, and Z-score normalized the genes across the cell lines to highlight differential expression across the lung cancer cell lines.
net.load_file('../lung_cellline_3_1_16/lung_cl_all_ptm/precalc_processed/CST_CCLE_exp.txt')
print('Expression data shape: ' + str(net.dat['mat'].shape))
Expression data shape: (1000, 37)
net.set_cat_color('row', 1, 'Data-Type: Exp', 'yellow')
net.cluster(views=[])
net.widget()
Cell Lines Cluster According to Histology and Mutation¶
Similarly to what we saw with the PTM data above, we see that cell lines again cluster according to their histology, sub-histology, and shared mutations. Again, we see that the cell line H2106 is the only NSCLC cell line that clusters with the SCLC cell lines. It is reassuring that we are seeing the same behavior for H2106 in these two independent datasets. We also see that cell lines cluster based on the RB1 mutation, but less so on KRAS and EGFR mutations. Cell lines may cluster based on EGFR mutation in PTM space and not gene-expression space since EGFR is a kinase that effects phosphorylation levels in PTM space.
We also see two high-level clusters of genes that have high/low expression in SCLC cell lines and vice versa in NSCLC cell lines.
# exp_sclc = net.widget_df()
# exp_nsclc = net.widget_df()
# exp_sclc.to_csv('histology_clusters/exp_sclc.txt', sep='\t')
# exp_nsclc.to_csv('histology_clusters/exp_nsclc.txt', sep='\t')
Merge PTM and Gene Expression Data¶
Above, we saw broadly similar patterns in lung cancer cell line behavior in PTM- and gene-expression-space:
- lung cancer cell lines cluster according to their histology, sub-histology, and shared mutations
- PTMs and genes form two large clusters with opposite expression in SCLC and NSCLC cell lines
Since we have processed the datasets similarly (e.g. Z-scored across cell lines) we can look for cross-data-type clusters by simply stacking our datasets vertically (see CST_Data_Processing.ipynb for more information). Clustering this mixed data will help us identify potentially important correlations between expression, phosphorylation, methylation, and acetylation (see CST_Data_Processing.ipynb for more information on merging the datasets).
# load merged PTM and gene expression data
net.load_file('../lung_cellline_3_1_16/lung_cl_all_ptm/precalc_processed/CST_CCLE_merge.txt')
net.cluster(views=[])
net.widget()
Cell Lines Cluster According to Histology and Mutation¶
As expected, we see that our cell lines again cluster according to their histology and mutations. We can now look at the two clusters of differentially regulated PTM/genes across NSCLC and SCLC cell lines. Below are the breakdowns of data-types in the two large clusters identified at the default (level 5) of the interactive dendrogram:
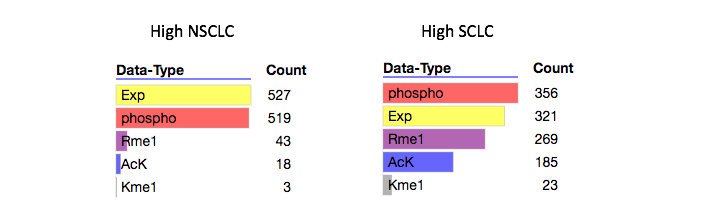
We see that the top cluster that is up-regulated in NSCLC cell lines is composed almost exclusively of gene expression data and phosphorylation data. The cluster that is up-regulated in SCLC cell lines is composed mostly of phosphorylation, expression, argining methylation, and lysine acetylation data.
Below and in the linked notebooks we will investigate the clusters of up-regulated PTMs/genes in SCLC and NSCLC cell lines.
# merge_sclc = net.widget_df()
# merge_nsclc = net.widget_df()
# merge_sclc.to_csv('histology_clusters/merge_sclc.txt', sep='\t')
# merge_nsclc.to_csv('histology_clusters/merge_nsclc.txt', sep='\t')
SCLC PTM and Gene-Expression Cluster¶
Here and in the notebok Merged_SCLC_Cluster.ipynb we investigate the cluster of up-regulated PTMs and expressed genes in SCLC cell lines. Below we are visualizing the cluster of up-regulated PTMs/expressed-genes in SCLC cell lines and pre-calculating Gene Ontology Biological Process enrichment.
net.load_file('histology_clusters/merge_sclc.txt')
net.enrichrgram('GO_Biological_Process_2015')
net.cluster(views=[])
net.widget()
mRNA Processing and Neuronal Functions¶
Using enrichment analysis for Gene Ontology Biological Function we see that the genes/proteins with up-regulated PTMs and expression levels are associated with RNA processing, RNA splicing and, gene expression. Re-running the analysis for the sub-cluster of very highly regulated PTMS/genes (the large cluster in the middle) reveals enrichment for neuronal functions including: neuron projection, axon guidance, and neuron morphology. Enrichment for genes that are up-regulated in disease (using the Disease Perturbations from GEO Up library) shows enrichment for neuronal related cancers including: oligodendroglioma, multiple sclerosis, astrocytoma, and large cell neuroendocrine carcinoma. Finally, enrichment using the MGI Mammalian Phenotype library shows enrichment for genes that cause neuronal abnormalities in mice after knockdown including: abnormal neuron morphology, abnormal brain morphology, abnormal spinal cord morphology, and abnormal nervous system. Collectively, these results indicate that the genes/proteins that co-cluster and have high PTM/gene-expression levels in SCLC cell lines have neuronal functions. This agrees with the neuronal characteristics of SCLC cell lines (see Onganar et al. 2005). For more information see the Merged_SCLC_cluster.ipynb notebook.
NKX2-1 Cluster¶
We find that the lung assocaited transcription factor NKX2-1 (which has been used as a biomarker in lung cancer, Yang et al. 2012) has similar gene expression and arginine methylation levels across lung cancer cell lines and is clustered with a set of cytoskeletal and extracellular matrix related proteins (e.g. actin, myosin). This might point to a functionally important role for NKX2-1 regulation of the cytoskeleton and ECM in lung cancer, which has been previously proposed Yamaguchi et al. 2013. For more information see the Merged_SCLC_cluster.ipynb notebook.
NSCLC PTM and Gene Expression Cluster¶
Here and in the notebook Merged_NSCLC_Cluster.ipynb we investigate the cluster of up-regulated PTMs and expressed genes in NSCLC cell lines. Below we are visualizing the cluster of up-regulated PTMs/expressed-genes in NSCLC cell lines and pre-calculating Gene Ontology Biological Process enrichment.
net.load_file('histology_clusters/merge_nsclc.txt')
net.enrichrgram('GO_Biological_Process_2015')
net.cluster(views=[])
net.widget()
Cell Motility¶
Using enrichment analysis for Gene Ontology Biological Function we see that genes/proteins with up-regulated PTMs and expression levels are associated with cell motility terms including: cellular component movement, cell motility, cell migration, locomotion, response to wound healing, and cell adhesion. This broadly agrees with prior knowledge that NSCLC cell lines are known to form adherent monolayers while SCLC cell lines grow in aggregates (Doyle et al. 1990). Similarly, enrichment results using the KEGG library show enrichment for focal adhesion among other enrichment results including proteoglycans in cancer and neurotrophin signaling.
Similarities to Other Cancers and Diseases¶
Enrichment using the Disease Perturbations from GEO up library shows enrichment for diseases including: pancreatic cancer and Barrett's esophagus. This might point to functional similarities between these disesases and NSCLC lung cancer.
Possible Immune Functions¶
Enrichment using the MGI Mammalian Phenotype library shows enrichment for several immune related phenotypes including: abnormal innate immunity and abnormal antigen presenting.
For more information see the Merged_NSCLC_cluster.ipynb notebook.