De novo Assembly¶
In [ ]:
Here are two files to be used for de novo assembly
In [6]:
#file filtered_106A_Female_Mix_GATCAG_L004_R1.fastq
from IPython.display import IFrame
IFrame('http://de.iplantcollaborative.org/dl/6ffe582a-6920-468a-a234-92893412c49a', width=1000, height=350)
Out[6]:
In [7]:
#file filtered_106A_Female_Mix_GATCAG_L004_R2.fastq
from IPython.display import IFrame
IFrame('http://de.iplantcollaborative.org/dl/d1ce64ee-4a53-4799-b93a-03c05eb4439b', width=1000, height=350)
Out[7]:
Running Trinity on Iplant¶
In [7]:
cd /Volumes/web/whale/fish546/
/Volumes/web/whale/fish546
In [10]:
from IPython.display import IFrame
IFrame('http://de.iplantcollaborative.org/dl/bcbac543-5778-4826-87d4-aadf6e4a1540', width=1000, height=350)
Out[10]:
In [11]:
pwd
Out[11]:
u'/Users/sr320/Dropbox/Steven/ipython_nb/fish546'
In [12]:
cd /Volumes/web/cnidarian/
/Volumes/web/cnidarian
In [14]:
!curl -o 'fish546_14_Trinity.fasta' 'http://de.iplantcollaborative.org/dl/d/bcbac543-5778-4826-87d4-aadf6e4a1540/Trinity.fasta'
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 157M 0 157M 0 0 239k 0 --:--:-- 0:11:12 --:--:-- 298k
count | sum_len | N50 | min_len | max_len | med_len | ave_len | sd_len | file
113679 | 136406223 | 2375 | 201 | 32089 | 572 |1200 | 1545 | Trinity.fasta
In [16]:
import pandas
In [20]:
%%file data.csv
count,sum_len,N50,min_len,max_len,med_len,ave_len,sd_len,file
113679,136406223,2375,201,32089,572,1200,1545,Trinity.fasta
Overwriting data.csv
In [21]:
df = pandas.read_csv('data.csv')
In [25]:
from IPython.display import HTML
In [ ]:
In [34]:
df.head
Out[34]:
<bound method DataFrame.head of count sum_len N50 min_len max_len med_len ave_len sd_len \
0 113679 136406223 2375 201 32089 572 1200 1545
file
0 Trinity.fasta >
In [36]:
cd Desktop/
/Users/sr320/Desktop
In [37]:
!perl count_fasta.pl
usage: count_fasta.pl [-i <interval size in # of residues>] <fasta file(s)> Produces a histogram of sequence lengths and other measures to standard out. -i # specify bin size for histogram (default 100)
In [40]:
!perl count_fasta.pl /Volumes/web/cnidarian/fish546_14_Trinity.fasta
200:299 29818 300:399 13977 400:499 8480 500:599 6072 600:699 4794 700:799 4036 800:899 3484 900:999 3054 1000:1099 2767 1100:1199 2498 1200:1299 2327 1300:1399 2165 1400:1499 2026 1500:1599 1895 1600:1699 1684 1700:1799 1518 1800:1899 1439 1900:1999 1289 2000:2099 1220 2100:2199 1097 2200:2299 1061 2300:2399 970 2400:2499 1017 2500:2599 880 2600:2699 865 2700:2799 792 2800:2899 717 2900:2999 652 3000:3099 601 3100:3199 609 3200:3299 554 3300:3399 513 3400:3499 415 3500:3599 477 3600:3699 410 3700:3799 415 3800:3899 365 3900:3999 393 4000:4099 320 4100:4199 371 4200:4299 268 4300:4399 251 4400:4499 257 4500:4599 253 4600:4699 286 4700:4799 251 4800:4899 184 4900:4999 148 5000:5099 181 5100:5199 171 5200:5299 197 5300:5399 143 5400:5499 163 5500:5599 140 5600:5699 186 5700:5799 134 5800:5899 157 5900:5999 110 6000:6099 104 6100:6199 84 6200:6299 99 6300:6399 97 6400:6499 83 6500:6599 65 6600:6699 80 6700:6799 59 6800:6899 74 6900:6999 68 7000:7099 91 7100:7199 53 7200:7299 81 7300:7399 50 7400:7499 56 7500:7599 62 7600:7699 62 7700:7799 48 7800:7899 36 7900:7999 33 8000:8099 40 8100:8199 33 8200:8299 14 8300:8399 15 8400:8499 30 8500:8599 48 8600:8699 36 8700:8799 44 8800:8899 36 8900:8999 14 9000:9099 10 9100:9199 8 9200:9299 15 9300:9399 11 9400:9499 28 9500:9599 15 9600:9699 7 9700:9799 12 9800:9899 8 9900:9999 5 10000:10099 8 10100:10199 8 10200:10299 4 10300:10399 5 10400:10499 6 10500:10599 13 10600:10699 8 10700:10799 6 10800:10899 7 10900:10999 1 11000:11099 6 11100:11199 9 11200:11299 3 11300:11399 0 11400:11499 5 11500:11599 5 11600:11699 13 11700:11799 0 11800:11899 1 11900:11999 6 12000:12099 4 12100:12199 3 12200:12299 5 12300:12399 3 12400:12499 10 12500:12599 7 12600:12699 13 12700:12799 6 12800:12899 7 12900:12999 15 13000:13099 7 13100:13199 2 13200:13299 2 13300:13399 0 13400:13499 1 13500:13599 9 13600:13699 5 13700:13799 8 13800:13899 2 13900:13999 7 14000:14099 5 14100:14199 4 14200:14299 2 14300:14399 0 14400:14499 0 14500:14599 3 14600:14699 3 14700:14799 7 14800:14899 5 14900:14999 5 15000:15099 4 15100:15199 6 15200:15299 5 15300:15399 0 15400:15499 0 15500:15599 8 15600:15699 2 15700:15799 3 15800:15899 1 15900:15999 0 16000:16099 1 16100:16199 1 16200:16299 0 16300:16399 0 16400:16499 0 16500:16599 2 16600:16699 0 16700:16799 0 16800:16899 1 16900:16999 0 17000:17099 0 17100:17199 1 17200:17299 8 17300:17399 10 17400:17499 13 17500:17599 3 17600:17699 0 17700:17799 0 17800:17899 0 17900:17999 1 18000:18099 0 18100:18199 0 18200:18299 0 18300:18399 0 18400:18499 5 18500:18599 0 18600:18699 0 18700:18799 0 18800:18899 4 18900:18999 0 19000:19099 0 19100:19199 0 19200:19299 0 19300:19399 0 19400:19499 0 19500:19599 0 19600:19699 0 19700:19799 0 19800:19899 0 19900:19999 0 20000:20099 0 20100:20199 0 20200:20299 0 20300:20399 0 20400:20499 0 20500:20599 0 20600:20699 0 20700:20799 0 20800:20899 0 20900:20999 2 21000:21099 1 21100:21199 0 21200:21299 0 21300:21399 0 21400:21499 0 21500:21599 0 21600:21699 0 21700:21799 0 21800:21899 0 21900:21999 0 22000:22099 0 22100:22199 0 22200:22299 0 22300:22399 0 22400:22499 0 22500:22599 0 22600:22699 0 22700:22799 0 22800:22899 0 22900:22999 0 23000:23099 0 23100:23199 0 23200:23299 0 23300:23399 0 23400:23499 0 23500:23599 0 23600:23699 0 23700:23799 0 23800:23899 0 23900:23999 0 24000:24099 0 24100:24199 0 24200:24299 0 24300:24399 0 24400:24499 0 24500:24599 0 24600:24699 0 24700:24799 0 24800:24899 0 24900:24999 0 25000:25099 0 25100:25199 0 25200:25299 0 25300:25399 0 25400:25499 0 25500:25599 0 25600:25699 0 25700:25799 0 25800:25899 0 25900:25999 0 26000:26099 0 26100:26199 0 26200:26299 0 26300:26399 0 26400:26499 0 26500:26599 0 26600:26699 0 26700:26799 0 26800:26899 0 26900:26999 0 27000:27099 0 27100:27199 0 27200:27299 0 27300:27399 0 27400:27499 0 27500:27599 0 27600:27699 0 27700:27799 0 27800:27899 0 27900:27999 0 28000:28099 0 28100:28199 0 28200:28299 0 28300:28399 0 28400:28499 0 28500:28599 0 28600:28699 0 28700:28799 0 28800:28899 0 28900:28999 0 29000:29099 0 29100:29199 0 29200:29299 0 29300:29399 0 29400:29499 0 29500:29599 0 29600:29699 0 29700:29799 0 29800:29899 0 29900:29999 0 30000:30099 0 30100:30199 0 30200:30299 0 30300:30399 0 30400:30499 0 30500:30599 0 30600:30699 0 30700:30799 0 30800:30899 0 30900:30999 0 31000:31099 0 31100:31199 0 31200:31299 0 31300:31399 0 31400:31499 0 31500:31599 0 31600:31699 0 31700:31799 0 31800:31899 0 31900:31999 0 32000:32099 2 Total length of sequence: 136406223 bp Total number of sequences: 113679 N25 stats: 25% of total sequence length is contained in the 5366 sequences >= 4305 bp N50 stats: 50% of total sequence length is contained in the 16227 sequences >= 2375 bp N75 stats: 75% of total sequence length is contained in the 37135 sequences >= 1102 bp Total GC count: 54597739 bp GC %: 40.03 %
In [115]:
from pandas import *
# read data from data file into a pandas DataFrame
hi = read_table("/Users/sr320/Desktop/histo.txt") # name of the data file
#sep=",", # what character separates each column?
#na_values=["", " "]) # what values should be considered "blank" values?
In [116]:
print hi
<class 'pandas.core.frame.DataFrame'>
Int64Index: 319 entries, 0 to 318
Data columns (total 2 columns):
INT 319 non-null values
count 319 non-null values
dtypes: int64(1), object(1)
In [117]:
hi
Out[117]:
<class 'pandas.core.frame.DataFrame'>
Int64Index: 319 entries, 0 to 318
Data columns (total 2 columns):
INT 319 non-null values
count 319 non-null values
dtypes: int64(1), object(1)
In [118]:
hi.head()
Out[118]:
| INT | count | |
|---|---|---|
| 0 | 200:299 | 29818 |
| 1 | 300:399 | 13977 |
| 2 | 400:499 | 8480 |
| 3 | 500:599 | 6072 |
| 4 | 600:699 | 4794 |
In [77]:
----
File "<ipython-input-77-404fb44d22bf>", line 1 ---- ^ SyntaxError: invalid syntax
In [78]:
!perl -e '$count=0; $len=0; while(<>) {s/\r?\n//; s/\t/ /g; if (s/^>//) { if ($. != 1) {print "\n"} s/ |$/\t/; $count++; $_ .= "\t";} else {s/ //g; $len += length($_)} print $_;} print "\n"; warn "\nConverted $count FASTA records in $. lines to tabular format\nTotal sequence length: $len\n\n";' /Volumes/web/cnidarian/fish546_14_Trinity.fasta > /Volumes/web/cnidarian/fish546_14_Trinity.tab
Converted 113679 FASTA records in 2442378 lines to tabular format Total sequence length: 136406223
In [99]:
!perl -e '$col = 2;' -e 'while (<>) { s/\r?\n//; @F = split /\t/, $_; $len = length($F[$col]); print "$_\t$len\n" } warn "\nAdded column with length of column $col for $. lines.\n\n";' /Volumes/web/cnidarian/fish546_14_Trinity.tab > /Volumes/web/cnidarian/fish546_14_Trinity_ls_length2.tab
Added column with length of column 2 for 113679 lines.
In [100]:
!head -2 /Volumes/web/cnidarian/fish546_14_Trinity_ls_length2.tab
comp0_c0_seq1 len=526 path=[4411:0-525] TCTGTGCCGATATATATATACATGTATCTATGCACAGTCATGTTTTATCGAGTACGACTGTAAAAAAGATCCAAAGTATACTCAAAAAATCTTCAGCAAGTGAGTCCATTTGAGTATGAGAATGATATTATCAAAGTTTTAAAACAATTTAGAGCATAGAGGTTGAGTCCGTGTATGAAAATGTGCTCAAAAAAGTAAATATAACTTCCAGGACAATTATTTTTACCGGCGTCTACTTGTATCTGTACGTATATTAAAAGTCTTAACGGCCACTGAACGAGCCAGCCTATTTATAAACTTGTTAACGCTTCCTTAAATGGCTGTAACTGAAGCTCCAAGCAAGTACTTAGCTAGAAAACACCCAGACGTACGTAAAAGTTTCCTTATTTACTGAAAATTTAAATGTATTCACCCAGTAGCACCCGGGGCCTTGATGAGGCCACGATAAATCTGCTTTTCAACAACCATAATGATTTGTTAATTCATAAGAAACACGCTCATGTATAGACTTCGATTTCAGGATAAG 526 comp1_c0_seq1 len=265 path=[1100:0-54 2691:55-264] GGGGAATTCAGCTAACATACCTCGATGATGAGGGGGAGCCGTCCTGTCAAAACTCCAATAATCTGAATTATACACTGCTGAAGCACTTAACACGTCAAGATGTGGGGTGTGCCTTAGATACAACATCTACAAAAATAAATCTAGACACTGACCTGGACTCCAGAGCCGATACAGACACTGTCCTTACTGAAATTACCAAACCTGGCAAGTATTGAACGTGTACCAAAACTTATAGATTTTGTATATGGTATTAATTTCTCTGGTG 265
In [101]:
from pandas import *
# read data from data file into a pandas DataFrame
la = read_table("http://eagle.fish.washington.edu/cnidarian/fish546_14_Trinity_ls_length2.tab", # name of the data file
#sep=",", # what character separates each column?
na_values=["", " "]) # what values should be considered "blank" values?
In [103]:
la.head()
Out[103]:
<class 'pandas.core.frame.DataFrame'> Int64Index: 5 entries, 0 to 4 Data columns (total 4 columns): comp0_c0_seq1 5 non-null values len=526 path=[4411:0-525] 5 non-null values TCTGTGCCGATATATATATACATGTATCTATGCACAGTCATGTTTTATCGAGTACGACTGTAAAAAAGATCCAAAGTATACTCAAAAAATCTTCAGCAAGTGAGTCCATTTGAGTATGAGAATGATATTATCAAAGTTTTAAAACAATTTAGAGCATAGAGGTTGAGTCCGTGTATGAAAATGTGCTCAAAAAAGTAAATATAACTTCCAGGACAATTATTTTTACCGGCGTCTACTTGTATCTGTACGTATATTAAAAGTCTTAACGGCCACTGAACGAGCCAGCCTATTTATAAACTTGTTAACGCTTCCTTAAATGGCTGTAACTGAAGCTCCAAGCAAGTACTTAGCTAGAAAACACCCAGACGTACGTAAAAGTTTCCTTATTTACTGAAAATTTAAATGTATTCACCCAGTAGCACCCGGGGCCTTGATGAGGCCACGATAAATCTGCTTTTCAACAACCATAATGATTTGTTAATTCATAAGAAACACGCTCATGTATAGACTTCGATTTCAGGATAAG 5 non-null values 526 5 non-null values dtypes: int64(1), object(3)
In [113]:
la ['526'].hist(bins=1000);
#Axis limits are changed using the axis([xmin, xmax, ymin, ymax]) function.
plt.axis([0, 4000, 0, 11000])
Out[113]:
[0, 4000, 0, 11000]

In [120]:
!perl /Users/sr320/Desktop/trinityrnaseq_r20131110/util/TrinityStats.pl /Volumes/web/cnidarian/fish546_14_Trinity.fasta
################################ ## Counts of transcripts, etc. ################################ Total trinity transcripts: 113679 Total trinity components: 69205 Percent GC: 40.03 ######################################## Stats based on ALL transcript contigs: ######################################## Contig N10: 6804 Contig N20: 4912 Contig N30: 3808 Contig N40: 2996 Contig N50: 2375 Median contig length: 572 Average contig: 1199.92 Total assembled bases: 136406223 ##################################################### ## Stats based on ONLY LONGEST ISOFORM per COMPONENT: ##################################################### Contig N10: 5174 Contig N20: 3631 Contig N30: 2726 Contig N40: 2099 Contig N50: 1604 Median contig length: 382 Average contig: 823.03 Total assembled bases: 56957585
Running CD-Hit on IPlant¶
In [2]:
cd /Volumes/web/cnidarian/
/Volumes/web/cnidarian
In [3]:
!curl -o 'fishfe6_oCD-HITout.fa' 'http://de.iplantcollaborative.org/dl/d/26cb3b68-ea18-4a9f-a723-247e03c0166a/CD-HITout.fa'
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 84.0M 0 84.0M 0 0 327k 0 --:--:-- 0:04:22 --:--:-- 332k
In [4]:
!head fishfe6_oCD-HITout.fa
>comp0_c0_seq1 len=526 path=[4411:0-525] TCTGTGCCGATATATATATACATGTATCTATGCACAGTCATGTTTTATCGAGTACGACTG TAAAAAAGATCCAAAGTATACTCAAAAAATCTTCAGCAAGTGAGTCCATTTGAGTATGAG AATGATATTATCAAAGTTTTAAAACAATTTAGAGCATAGAGGTTGAGTCCGTGTATGAAA ATGTGCTCAAAAAAGTAAATATAACTTCCAGGACAATTATTTTTACCGGCGTCTACTTGT ATCTGTACGTATATTAAAAGTCTTAACGGCCACTGAACGAGCCAGCCTATTTATAAACTT GTTAACGCTTCCTTAAATGGCTGTAACTGAAGCTCCAAGCAAGTACTTAGCTAGAAAACA CCCAGACGTACGTAAAAGTTTCCTTATTTACTGAAAATTTAAATGTATTCACCCAGTAGC ACCCGGGGCCTTGATGAGGCCACGATAAATCTGCTTTTCAACAACCATAATGATTTGTTA ATTCATAAGAAACACGCTCATGTATAGACTTCGATTTCAGGATAAG
In [5]:
!cat fishfe6_oCD-HITout.fa | awk '((NR-2)%4==0){read=$1;total++;count[read]++}END{for(read in count){if(!max||count[read]>max) {max=count[read];maxRead=read};if(count[read]==1){unique++}};print total,unique,unique*100/total,maxRead,count[maxRead],count[maxRead]*100/total}'
340785 321619 94.3759 G 126 0.0369735
In [6]:
!fgrep -c ">" fishfe6_oCD-HITout.fa
81885
Bowtie on IPlant to get alignment¶
In [10]:
!head BowtieOut.sam
^C
In [13]:
!/Volumes/Bay3/Software/BSMAP/bsmap-2.74/samtools/samtools -help
[main] unrecognized command '-help'
Tophat on IPlant to get alignment¶
In [ ]:
Visualized in IGV
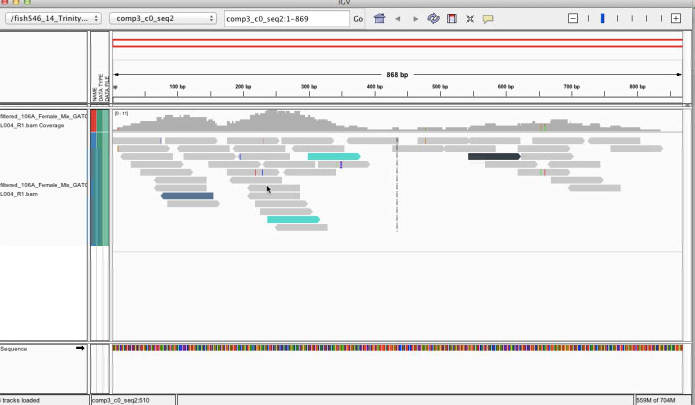
mpileup on IPlant¶
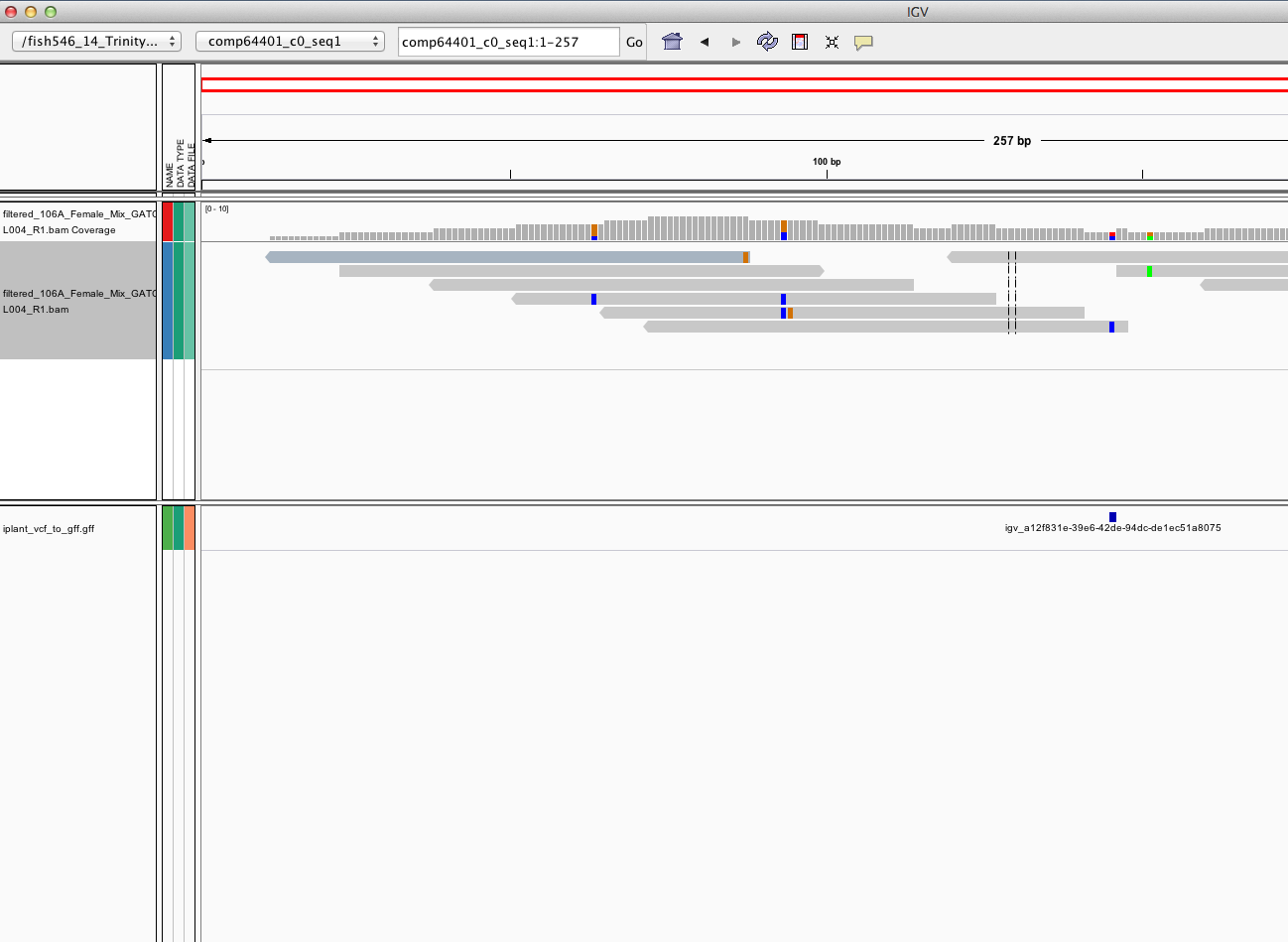
-- Trinity on hummingbird¶
In [3]:
!/usr/local/bioinformatics/trinity/Trinity.pl
###############################################################################
#
# ______ ____ ____ ____ ____ ______ __ __
# | || \ | || \ | || || | |
# | || D ) | | | _ | | | | || | |
# |_| |_|| / | | | | | | | |_| |_|| ~ |
# | | | \ | | | | | | | | | |___, |
# | | | . \ | | | | | | | | | | |
# |__| |__|\_||____||__|__||____| |__| |____/
#
###############################################################################
#
# Required:
#
# --seqType <string> :type of reads: ( fa, or fq )
#
# --JM <string> :(Jellyfish Memory) number of GB of system memory to use for
# k-mer counting by jellyfish (eg. 10G) *include the 'G' char
#
# If paired reads:
# --left <string> :left reads, one or more (separated by space)
# --right <string> :right reads, one or more (separated by space)
#
# Or, if unpaired reads:
# --single <string> :single reads, one or more (note, if single file contains pairs, can use flag: --run_as_paired )
#
####################################
## Misc: #########################
#
# --SS_lib_type <string> :Strand-specific RNA-Seq read orientation.
# if paired: RF or FR,
# if single: F or R. (dUTP method = RF)
# See web documentation.
#
# --output <string> :name of directory for output (will be
# created if it doesn't already exist)
# default( "/Users/steven/Dropbox/Steven/ipython_nb/fish546/trinity_out_dir" )
# --CPU <int> :number of CPUs to use, default: 2
# --min_contig_length <int> :minimum assembled contig length to report
# (def=200)
# --genome_guided :set to genome guided mode, only retains assembly fasta file.
# --jaccard_clip :option, set if you have paired reads and
# you expect high gene density with UTR
# overlap (use FASTQ input file format
# for reads).
# (note: jaccard_clip is an expensive
# operation, so avoid using it unless
# necessary due to finding excessive fusion
# transcripts w/o it.)
#
# --prep :Only prepare files (high I/O usage) and stop before kmer counting.
#
# --no_cleanup :retain all intermediate input files.
# --full_cleanup :only retain the Trinity fasta file, rename as ${output_dir}.Trinity.fasta
#
# --cite :show the Trinity literature citation
#
# --version :reports Trinity version (Trinityrnaseq_r20131110) and exits.
#
####################################################
# Inchworm and K-mer counting-related options: #####
#
# --min_kmer_cov <int> :min count for K-mers to be assembled by
# Inchworm (default: 1)
# --inchworm_cpu <int> :number of CPUs to use for Inchworm, default is min(6, --CPU option)
#
# --no_run_inchworm :stop after running jellyfish, before inchworm.
#
###################################
# Chrysalis-related options: ######
#
# --max_reads_per_graph <int> :maximum number of reads to anchor within
# a single graph (default: 200000)
# --no_run_chrysalis :stop Trinity after Inchworm and before
# running Chrysalis
# --no_run_quantifygraph :stop Trinity just before running the
# parallel QuantifyGraph computes, to
# leverage a compute farm and massively
# parallel execution..
#
# --chrysalis_output <string> :name of directory for chrysalis output (will be
# created if it doesn't already exist)
# default( "chrysalis" )
#
# --no_bowtie :dont run bowtie to use pair info in chrysalis clustering.
#
#####################################
### Butterfly-related options: ####
#
# --bfly_opts <string> :additional parameters to pass through to butterfly
# (see butterfly options: java -jar Butterfly.jar ).
# (note: only for expert or experimental use. Commonly used parameters are exposed through this Trinity menu here).
#
# //////////////////////////////////
# Alternative reconstruction modes:
# Default mode is the 'regular' Butterfly transcript reconstruction by graph node extension.
#
# --PasaFly PASA-like algorithm for maximally-supported isoforms (conservative reconstructions, fewer isoforms)
# or
# --CuffFly Cufflinks-like algorithm to report minimum transcripts (fewest isoforms)
#
#
# Butterfly read-pair grouping settings (used for all reconstruction modes to define 'pair paths'):
#
# --group_pairs_distance <int> :maximum length expected between fragment pairs (default: 500)
# (reads outside this distance are treated as single-end)
#
# ///////////////////////////////////////////////
# Butterfly default reconstruction mode settings. (no CuffFly or PasaFly custom settings are currently available).
#
# --path_reinforcement_distance <int> :minimum overlap of reads with growing transcript
# path (default: PE: 75, SE: 25)
# Set to 1 for the most lenient path extension requirements.
#
# --triplet_lock : (increase stringency of regular butterfly reconstruction)
# lock triplet-supported nodes: node 'c' having read path 'A-B-C' disables 'Z-B-C' if no such read support exists.
#
# --extended_lock : (further increase the stringency of regular butterfy reconstruction)
# extend the triplet lock to include longer range read path information.
# ex. in extending path 'A-B-Z' to 'A-B-Z-D', we only find read support for 'A-B-C-D', that 'A-B-Z' extension to 'D' will be blocked.
# (assumes --triplet_lock)
#
# /////////////////////////////////////////
# Butterfly transcript reduction settings:
#
# --no_path_merging : all transcript candidates are output (including SNP variations, however, some SNPs may be unphased)
#
# By default, alternative transcript candidates are merged (in reality, discarded) if they are found to be too similar, according to the following logic:
#
# (identity=(numberOfMatches/shorterLen) > 95.0% or if we have <= 2 mismatches) and if we have internal gap lengths <= 10
#
# with parameters as:
#
# --min_per_id_same_path <int> default: 95 min percent identity for two paths to be merged into single paths
# --max_diffs_same_path <int> default: 2 max allowed differences encountered between path sequences to combine them
# --max_internal_gap_same_path <int> default: 10 maximum number of internal consecutive gap characters allowed for paths to be merged into single paths.
#
# If, in a comparison between two alternative transcripts, they are found too similar, the transcript with the greatest cumulative
# compatible read (pair-path) support is retained, and the other is discarded.
#
#
# //////////////////////////////////////////////
# Butterfly Java and parallel execution settings.
#
# --bflyHeapSpaceMax <string> :java max heap space setting for butterfly
# (default: 10G) => yields command
# 'java -Xmx10G -jar Butterfly.jar ... $bfly_opts'
# --bflyHeapSpaceInit <string> :java initial hap space settings for
# butterfly (default: 1G) => yields command
# 'java -Xms1G -jar Butterfly.jar ... $bfly_opts'
# --bflyGCThreads <int> :threads for garbage collection
# (default, not specified, so java decides)
# --bflyCPU <int> :CPUs to use (default will be normal
# number of CPUs; e.g., 2)
# --bflyCalculateCPU :Calculate CPUs based on 80% of max_memory
# divided by maxbflyHeapSpaceMax
# --no_run_butterfly :stops after the Chrysalis stage. You'll
# need to run the Butterfly computes
# separately, such as on a computing grid.
# Then, concatenate all the Butterfly assemblies by running:
# 'find trinity_out_dir/ -name "*allProbPaths.fasta"
# -exec cat {} + > trinity_out_dir/Trinity.fasta'
#
#################################
# Grid-computing options: #######
#
# --grid_computing_module <string> : Perl module in /usr/local/bioinformatics/trinityrnaseq_r20131110/PerlLibAdaptors/
# that implements 'run_on_grid()'
# for naively parallel cmds. (eg. 'BroadInstGridRunner')
#
#
###############################################################################
#
# *Note, a typical Trinity command might be:
# Trinity.pl --seqType fq --JM 100G --left reads_1.fq --right reads_2.fq --CPU 6
#
# see: /usr/local/bioinformatics/trinityrnaseq_r20131110/sample_data/test_Trinity_Assembly/
# for sample data and 'runMe.sh' for example Trinity execution
# For more details, visit: http://trinityrnaseq.sf.net
#
###############################################################################
In [2]:
cd /Volumes/Data/sr320_scratch/
/Volumes/Data/sr320_scratch
In [3]:
pwd
Out[3]:
u'/Volumes/Data/sr320_scratch'
In [ ]: