Immune Cell Phosphorylation Response to PMA¶
White blood cells are a key component of the immune system and kinase signaling is known to play an important role in immune cell function (see Isakov and Altman 2013). Our collaborators at the Icahn School of Medicine Human Immune Monitoring Core used Mass cytometry, CyTOF (Fluidigm), to investigate the phosphporylation response of peripheral blood mononuclear cells (PBMC) immune cells exposed to PMA (phorbol 12-myristate 13-acetate), a tumor promoter and activator of protein kinase C (PKC) (see Wiki).
A total of 28 markers (18 surface markers and 10 phosphorylation markers) were measured in over 200,000 single cells. In this series of notebooks, we will semi-automatically identify cell types using surface markers and cluster single cells based on phosphorylation to identify cell-type specific behavior at the phosphorylation level. This proof of concept will demonstrate how Clustergrammer can be an effective means to visualize and analyze high-dimensional CyTOF data.
Load Data and Clustergrammer-Widget¶
First, we will instantiate an instance of the the Clustergrammer-Widget class that will be used to load, analyze, and visualize our data. For more information about using Clustergrammer's Network object to pre-process and cluster data see Clustergrammer-PY's API.
import pandas as pd
from clustergrammer_widget import *
net = Network(clustergrammer_widget)
Here will load the CyTOF datasets: PMA-treated (PMA) and not-PMA-treated (Plasma) (Note, we have slightly modified the format of our original CyTOF data to comply with Clustergrammer's data-formats using the precalc_cytof.py script). After loading our data, we select equal sized subsets (110,000 single cells) from each datasets. Then we combine our equal sized datasets to the DataFrame df_merge_ini, with 220,000 rows (110,000 plasma cells and 110,000 PMA cells) and 28 columns (18 surface markers and 10 phospho markers).
# Load Original Data and Subsample 110,000 Cells
################################################
net.load_file('../cytof_data/Plasma_clean.txt')
net.random_sample(axis='row', num_samples=110000, random_state=99)
df_plasma_ini = net.export_df()
net.load_file('../cytof_data/PMA_clean.txt')
net.random_sample(axis='row', num_samples=110000, random_state=99)
df_pma_ini = net.export_df()
# merge datasets and print shape of matrix
df_merge_ini = pd.concat([df_plasma_ini, df_pma_ini])
print(df_merge_ini.shape)
(220000, 28)
Plasma and PMA Treated with All Markers¶
For our first visualization, we will simply visualize a downsampled version of the Plasma and PMA treated cells using all markers to obtain a global overview (see Interacting with the Visualization for more information). This view will allow us to check some of our starting assumptions, such as: 1) we expect markers (columns) to cluster separately based on marker-type (e.g. surface and phospho markers), 2) we expect cells (rows) to cluster based on treatment (e.g. PMA treatment).
Before we visualize our data, we normalize the markers (Z-score the columns) so that their distributions are comparable, downsample the data using K-means to obtain 2000 cell-clusters and clip the Z-scored values at 10 standard deviations since we do not care about extreme outliers.
K-means clustering groups similar cells together, which can help us preserve rare cell populations while preventing large and homogeneous cell populations from overpowering the visualization. The size of each cluster is shown as the category number in clust with a color gradient - the darker the color the more cells are in the K-means identified cluster.
# load the merged dataframe the Clustergrammer's net object
net.load_df(df_merge_ini)
# manually set category colors
net.set_cat_color('col', 1, 'Marker-type: phospho marker', 'red')
net.set_cat_color('col', 1, 'Marker-type: surface marker', 'blue')
net.set_cat_color('row', 1, 'Majority-Treatment: Plasma', 'blue')
net.set_cat_color('row', 1, 'Majority-Treatment: PMA', 'red')
# normalize columns
net.normalize(axis='col', norm_type='zscore', keep_orig=False)
# downsample cell (rows)
ds_data = net.downsample(ds_type='kmeans', axis='row', num_samples=2000)
# clip values
net.clip(-10, 10)
# cluster (using default parameters) and visualize widget
net.cluster(views=[])
net.widget()
/Users/nickfernandez/anaconda/lib/python2.7/site-packages/sklearn/cluster/k_means_.py:1382: RuntimeWarning: init_size=300 should be larger than k=2000. Setting it to 3*k init_size=init_size)
Phospho vs Surface Markers¶
As expected, we see phosphorylation markers (red category columns) for the most part cluster separately from surface markers (blue category columns). However, there are a few exceptions. The surface marker CD14 clusters more closely with phosphorylation markers and the phosphohrylation markers IKBa and pSTAT3 cluster more closely with surface markers. This implies that CD14 may be informative of phosphorylation status, while IKBa and pSTAT3 may be informative of cell-type status.
PMA Treatment¶
We see that cell-clusters (from K-means downsampling) are clustered into PMA-treated (red category rows) and non-treatment clusters (blue category rows). We also see distinct populations of cells defined by their distinctive surface-marker and phosphorylation composition.
We can obtain a high-level overview of the effects of PMA treatment on marker levels by double-clicking the 'Majority-Treatment' row category label to reorder based on this category. This reorders the rows based on their 'Majority-Tratment' category and shows that PMA treatment has its most prominent effects on phosphorylation and specifically the markers:
- pCREB
- pMAPKAP2
- pERK1 2
- pp38
These phosphorylation marker have higher levels after PMA treatment.
Analyze Surface Marker and Phosphorylation Marker Data Separately¶
While it is useful to visualize surface and phosphorylation data together, we will separate surface-marker and phospho-parmer data to obtain a more in-depth analysis. Below, we will use surface marker data to identify cell type and then overlay cell type onto phosphorylation marker data to associate cell type with phosphorylation status.
Cell Type Identification using Surface Markers¶
In the notebook, Generate_Cell_Types.ipynb, we semi-automatically identify cell types using hierarchical clustering of surface-marker data. First, we downsampled the Plasma (control untreated) and PMA datasets to 1000 clusters each using K-means clustering. Second, we merged the two datasets together, performed hierarchical clustering, and labeled cell-clusters based on thier hierarchical clustering (resulting in 27 clusters). Third, we averaged the surface marker levels for each of the 27 surface marker cell-clusters and our collaborators at the Icahn School of Medicine Human Immune Monitoring Core manually lableled each surface-marker cluster, which is shown below (for more information please see the Generate_Cell_Types.ipynb notebook). Note that the visualization below does not include the number of cells or the breakdown of Plasma vs PMA treated cells in each surface-marker cluster.
net.load_file('../cytof_data/plasma_pma_dendro_avg_SM.txt')
net.cluster()
net.widget()
Finally, this cell type information (based on surface-marker clustering) was transferred back to the original single cell data and saved to the following TSV files: Plasma_UCT.txt and PMA_UCT.txt (UCT stands for 'Updated Cell Types'). This single-cell cell-type labeled data was used in this notebook to visualize and analyze cell-type specific phosphorylation behavior in response to PMA treatment.
Load Cell-Type Labeled Data¶
Here, we load the single-cell cell-type labeled data (discussed above):
# Cell-Type Labeled Data
########################
net.load_file('../cytof_data/Plasma_UCT.txt')
net.random_sample(axis='row', num_samples=110000, random_state=99)
df_plasma = net.export_df()
net.load_file('../cytof_data/PMA_UCT.txt')
net.random_sample(axis='row', num_samples=110000, random_state=99)
df_pma = net.export_df()
df_merge = pd.concat([df_plasma, df_pma])
print(df_merge.shape)
(220000, 28)
Set Cell Type Colors¶
Here we manually set the cell-type colors so they will be consistent across all visualizations. We have 16 unique cell types defined.
# manually set row colors: downsample
net.set_cat_color('row', 2, 'Majority-Category: B cells', '#22316C')
net.set_cat_color('row', 2, 'Majority-Category: Basophils', '#000033')
net.set_cat_color('row', 2, 'Majority-Category: CD14hi monocytes', 'yellow')
net.set_cat_color('row', 2, 'Majority-Category: CD14low monocytes', '#93b8bf')
net.set_cat_color('row', 2, 'Majority-Category: CD1c DCs', '#3636e2')
net.set_cat_color('row', 2, 'Majority-Category: CD4 Tcells', 'blue')
net.set_cat_color('row', 2, 'Majority-Category: CD4 Tcells_CD127hi', '#FF6347')
net.set_cat_color('row', 2, 'Majority-Category: CD4 Tcells CD161hi', '#F87531')
net.set_cat_color('row', 2, 'Majority-Category: CD4 Tcells_Tregs', '#8B4513')
net.set_cat_color('row', 2, 'Majority-Category: CD4 Tcells+CD27hi', '#330303')
net.set_cat_color('row', 2, 'Majority-Category: CD8 Tcells', '#ffb247')
net.set_cat_color('row', 2, 'Majority-Category: Neutrophils', 'purple')
net.set_cat_color('row', 2, 'Majority-Category: NK cells_CD16hi', 'red')
net.set_cat_color('row', 2, 'Majority-Category: NK cells_CD16hi_CD57hi', 'orange')
net.set_cat_color('row', 2, 'Majority-Category: NK cells_CD56hi', '#e052e5')
net.set_cat_color('row', 2, 'Majority-Category: Undefined', 'gray')
# manually set row colors: subsample
net.set_cat_color('row', 2, 'B cells', '#22316C')
net.set_cat_color('row', 2, 'Basophils', '#000033')
net.set_cat_color('row', 2, 'CD14hi monocytes', 'yellow')
net.set_cat_color('row', 2, 'CD14low monocytes', '#93b8bf')
net.set_cat_color('row', 2, 'CD1c DCs', '#3636e2')
net.set_cat_color('row', 2, 'CD4 Tcells', 'blue')
net.set_cat_color('row', 2, 'CD4 Tcells_CD127hi', '#FF6347')
net.set_cat_color('row', 2, 'CD4 Tcells CD161hi', '#F87531')
net.set_cat_color('row', 2, 'CD4 Tcells_Tregs', '#8B4513')
net.set_cat_color('row', 2, 'CD4 Tcells+CD27hi', '#330303')
net.set_cat_color('row', 2, 'CD8 Tcells', '#ffb247')
net.set_cat_color('row', 2, 'Neutrophils', 'purple')
net.set_cat_color('row', 2, 'NK cells_CD16hi', 'red')
net.set_cat_color('row', 2, 'NK cells_CD16hi_CD57hi', 'orange')
net.set_cat_color('row', 2, 'NK cells_CD56hi', '#e052e5')
net.set_cat_color('row', 2, 'Undefined', 'gray')
net.set_cat_color('row', 1, 'Treatment: Plasma', 'blue')
net.set_cat_color('row', 1, 'Treatment: PMA', 'red')
Plasma vs PMA Phosphorylation Subsample View¶
Since we cannot directly visualize a 220,000 row matrix using Clustergrammer we will take two approaches to visualize the data: subsampling and downsampling. First, we will use subsampling, which randomly selects 2000 cells out of the 220,000 cells from our combined dataset. Each visualization of the data will normalize phosphorylation marker distributions using Z-score and clip Z-score values at +-10 to disregard outliers.
# load PMA-treated and non-treated 'merged' data
net.load_df(df_merge)
# select only phospho markers
net.filter_cat('col', 1, 'Marker-type: phospho marker')
# Z-score normalize phosphorylation marker levels across all 200,000 single cells
net.normalize(axis='col', norm_type='zscore', keep_orig=False)
# randomly select 2000 cells
net.random_sample(axis='row', num_samples=2000, random_state=99)
# clip values (we do not care about extreme outliers)
net.clip(-10, 10)
# calculate clustering (with no row filtering views) and generate the widget
net.cluster(views=[])
net.widget()
PMA Effects on Phosphorylation Visualized using Subsampling¶
Above we see that we have a roughly equal number of Plasma and PMA treated cells, which is expected since we randomly selected cells from our combined dataset. We also see that PMA treated cells tend to cluster separately from Plasma treated cells regardless of cell type. From the bottom cluster of PMA treated cells it is clear that PMA treatment increases the following phosphorylations:
- pCREB
- pMAPKAP2
- pERK1 2
- pCREB
Cell-Clusters Identified using Subsampling¶
At level 5 of the row dendrogram (the default level, see Interactive Dendrogram) we see three large clusters. These clusters primarily consist of the following cell types (mouseover the dendrogram trapezoids to see a breakdown of category types in a cluster):
- Cell-Cluster 1: CD14hi monocytes, NK cells_CD16hi, CD4 Tcells
- Cell-Cluster 2: CD4 Tcells, CD8 Tcells, CD4 Tcells+CD27hi
- Cell-Cluster 3: CD4 Tcells, CD8 Tcells, CD14hi monocytes
Phospohrylation Clusters Identified using Subsampling¶
At level 6 of the column dendrogram we see two large clusters of phosphorylations:
- Phospho-Cluster 1: pPLCg2, pSTAT1, pSTAT5
- Phospho-Cluster 2: pCREB, pMAPKAP2, pERK1 2, pp38, pS6
IkBa appears to be uniquely regulated and pSTAT3 does not show much variation in its levels across different cells. Recall from first visualization that IkBa clusters more closely with surface markers than other phosphorylations.
Cell Type Clustering using Subsampling¶
There is some clustering based on cell type and the most obvious is the cluster of CD14hi monocytes (yellow) cells at the bottom of the heatmap with high levels of the above four phosphorylations. To more easily visualze cell-type phosphorylation behavior we can reorder cells based on their cell-type by double-clicking the 'Category 2' label on the top left of the clustergram. This arranges cells based on their cell type and allows us to see a few broad patterns:
- CD8 Tcells have high phosphorylation of IkBa
- NK cells CD16hi have high levels of pPLCg2, pSTAT1, and pSTAT5 and low levels of IkBa
We can also find associations between cell types and specific phosphorylations. For instance, reordering the rows based on pCREB levels shows that CD14hi monocytes are among the cells with the highest pCREB4 levels.
Plasma vs PMA Phosphorylation Downsample View¶
Randomly subsampling our data is a useful way to obtain a visualization of this large dataset, but subsampling may cause us to miss rare populations in our data. As discussed earlier, K-means clustering can be used as a means to downsample our data that can preserve rare populations as well as reduce the representation of large homogeneous populations. Below we have performed K-means clustering with 2000 clusters.
net.load_df(df_merge)
net.filter_cat('col', 1, 'Marker-type: phospho marker')
net.normalize(axis='col', norm_type='zscore', keep_orig=False)
ds_data = net.downsample(ds_type='kmeans', axis='row', num_samples=2000)
net.clip(-10, 10)
net.cluster(views=[])
net.widget()
Above we see a downsampled view of PMA-treated and non-treated single cell data as 2000 cell-clusters hierarchically clustered based on phosphorylation. Each cluster is assigned categorical information based on the categories of the majority of the cells in the cluster: e.g. Majority-Treatment and Majority-Category (cell type). Clusters vary in size from 1 cell to 531 cells.
PMA Effects on Phosphorylation Visualized using Downsampling¶
We see that PMA treated cells cluster separately from Plasma cells (red and blue row categories) and that we have obtained 1150 PMA clusters and only 850 Plasma cell line clusters. Since homogeneous populations are should be merged into large clusters with K-means clustering, this breakdown implies that Plasma treated cells may be more homogeneous in phosphorylation space. This might be expected since PMA treatment is expected initiate kinase signaling and thereby can be expected to create more heterogeneity in phosphorylation levels.
Cell-Clusters Identified using Downsampling¶
At level 5 of the row dendrogram (the default level) we see three large clusters. These clusters primarily consist of the following cell types:
- Cell-Cluster 1: CD4 Tcells, CD8 Tcells, NK cells CD16hi_CD57hi
- Cell-Cluster 2: NK cells_CD16hi, CD14hi monocytes, CD4 Tcells
- Cell-Cluster 3: CD4 Tcells, CD14hi monocytes, CD8 Tcells
Cell-Cluster 1 identified with downsampling corresponds to Cell Cluster 2 identified with subsampling: both have high populations of CD4Tcells and CD8 Tcells and have high phosphorylation of IKBA.
Cell-Cluster 2 identified with downsampling corresponds to Cell Cluster 1 identified with subsampling: both have high populations of NK cells_CD16hi and CD14hi monocytes with high phosphorylation levels of pPLCg2, pSTAT1, and pSTAT5.
Cell-Cluster 3 identified with downsampling corresponds to Cell Cluster 3 identified with subsampling: both have high populations of CD4 Tcells, CD8 Tcells, and CD14hi monocytes and have high phosphorylation of pCREB, pMAPKAP2, pERK1 2, pp38, and pS6.
Phospohrylation Clusters Identified using Downsampling¶
At level 7 of the column dendrogram we see two large clusters of phosphorylations:
- Phospho-Cluster 1: pCREB, pMAPKAP2, pERK1 2, pp38, pS6
- Phospho-Cluster 2: pPLCg2, pSTAT1, pSTAT5
Again, IkBa appears to be a uniquely regulated phosphorylation and pSTAT3 does not show much variation in its levels across different cells.
CD14hi Monocytes¶
PMA treated CD14hi monocytes (yellow row category) form one of the most prominent clusters in phosphorylation space with high levels of:
- pCREB
- pMAPKAP2
- pp38
- pERK1 2
NK cells CD16hi¶
NK cells CD16hi (red row category) cluster together (middle cluster). This cluster appears to be evenly composed of PMA and Plasma cells (see interactive dendrogram). These cells have relatively high phosphorylation levels of:
- pPLCg2
- pSTAT1
- pSTAT5
CD1c DCs¶
CD1c DCs (blue/purple row category) make up a small number of the cell-clusters in the downsampled data and their clustering is not clear when rows are in cluster order. However, reordering the rows based on their 'Majority-Category' by double-clicking this title (top left of the clustergram) shows that CD1c DCs have particularly high phosphorylation levels of pCREB, pMAPKAP2, pp38 and pERK1 2 when exposed to PMA (see red row category). We can also see these cell-clusters are relatively small (~10-60 single cells each) from the opacity of the 'number in clust' category. This might explain why the behavior of CD1c DCs was not as prominent with the subsampled data reordered based on cell-type category and demonstrates the ability of downsampling to highlight rare populations of cells with unique behavior.
Subsampling vs Downsampling¶
In the above two heatmaps, we see broadly similar clustering of cell-types and phosphorylations when we subsample or downsample. However, it is clear that there are differences in the cell-type distributions using the two methods, e.g. there are more yellow CD14hi monocytes with downsampling than with subsampling. We can get an overview of the breakdowns of cell-types in the datasets using the interactive dendrogram: increasing the size of the row dendrogram groups to the maximum so that all rows are in one group and then mousing eover or clicking the denrogram trapezoid gives a bar-graph breakdown of the categories.
Below are the breakdowns from subsampling and downsampling. We see that the diversity of cell types in the downsampled data is lower in the downsampled data vs the subsampled data. This might imply that phosphorylation data is not strongly dependent on all cell types.
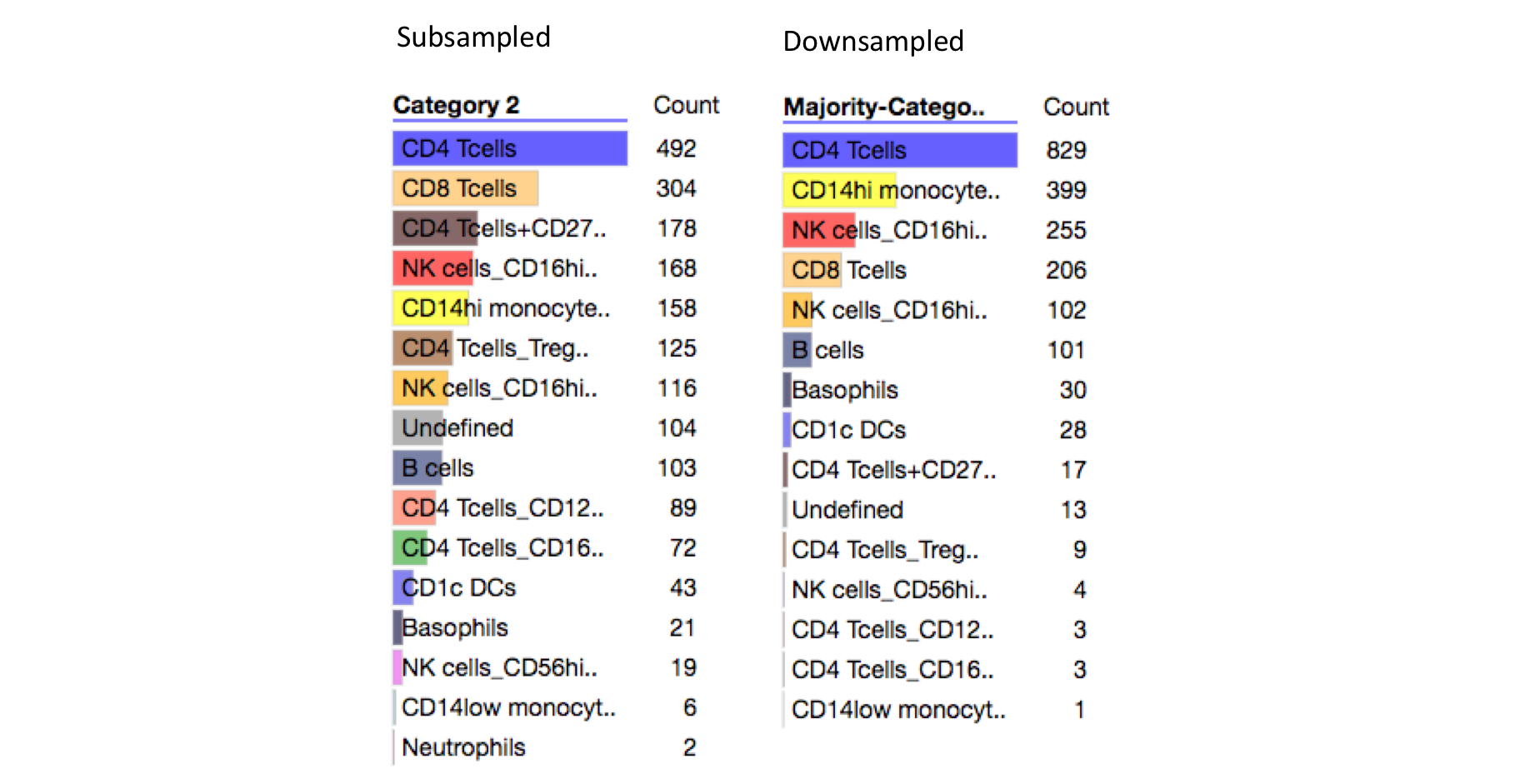
Downsampling Underrepresents CD8 Tcells and CD4 Tcells+CD27hi¶
The two cell types that are most underrepresented in the downsampled data are CD8 Tcells (2nd most common cell type in subsampled data) and CD4 Tcells+CD27hi (3rd most common cell type in subsampled data). We can see from the subsampled heatmap above that these cell lines are largely uniformly distributed in the heatmap, which might explain why they rarely make up the majority cell type in a K-means cluster.
Downsampling Overrepresents CD14hi monocytes¶
The most over-represented cell type in the downsampled data is CD14hi monocytes (increased from ~8% in subsampled data to ~20% in downsampled data). We can also see from the downsampled heatmap CD14hi monocytes cell clusters are relatively small (have a small number of cells, see 'number in clust' category).
This implies that: 1) CD14hi monocytes cell may cluster closely together in phosphorylation space (which appears to be the case from the subsampled heatmap) and 2) CD14hi monocytes have heterogeneous phosphorylation behavior that prevents them from being clustered into large homogeneous K-means clusters.
To aid visualization of this cell type we will generate a heatmap with only CD14hi monocytes cell clusters below:
net.filter_cat('row', 2, 'Majority-Category: CD14hi monocytes')
net.cluster(views=[])
net.widget()
Above we see that Plasma and PMA treated cell-clusters form two large clusters. These clusters are largely defined based on the four phosphorylations discussed above.
Additional Views¶
In this notebook we focused on PMA treatment's effects on phosphorylation levels. However, we can obtain additional views of the data by including Surface Markers or focusing on Plasma and PMA treated cells separately. Below are some links to additional notebooks take take alternative views of the data:
Plasma vs PMA Surface Markers¶
The notebook Plasma_vs_PMA_Surface_Markers.ipynb visualizes Plasma and PMA surface marker data. This notebook shows that PMA treated cells do not cluster separately based on surface markers, which is expected. The notebook also demonstrates how downsampling surface marker data increases the relative representation of rare cell types (e.g. Basophils) while dimishing the representation of dominant cell types (e.g. CD4 Tcells).
Plasma and PMA Treatment Separately Analyzed¶
The notebooks Plasma_Treatment.ipynb and PMA_Treatment visualize these datasets separately.
Conclusions¶
This notebook demonstrates that interactive heatmaps, produced using Clustergrammer, can be used to identify cell-type specific behavior in CyTOF data.