flotilla: Data-driven conversations¶
Open-source Python package for iterative machine learning and visualization | YeoLab/flotilla
Olga Botvinnik | @olgabot | github.com/olgabot | olgabotvinnik.com
PhD Candidate in Bioinformatics at UCSD | Gene Yeo Lab
National Defense Science and Engineering Graduate Fellow
NumFOCUS John Hunter Technical Fellow
April 10th, 2015

The time between computational idea to experimental result¶
What is flotilla?¶
flotilla is an open-source Python package for exploring data*.
*Currently, flotilla is focused on biological data such as single-cell and other large-scale RNA-seq transcriptome analyses
- Data
- Computation
- Visualization
- Iterativity

flotilla is ... Visualization¶
matplotlib: Robust plotting packageseaborn: Statistical data visualization
![]()



Why not just use these individual packages?¶
While individually, scikit-learn makes it easy to run individual algorithms, pandas makes subsetting data a dream, matplotlib and seaborn make visualizing computational results a charm, and the IPython notebook makes stringing all of these together into reproducible document possible, flotilla does something none of these other packages can.
flotilla shortens the distance between a hypothesis and its computational result¶
Hypothesis: The human brain uses different genes in different regions¶
The data: Allen Brain Institute¶
Disclaimer: I am not a neuroscientist, and my understanding of brain anatomy is very rudimentary, so please bear with me.
We will use the BrainSpan Atlas of the Developing Human Brain, which was an effort to establish molecular profiles of brain regions at varying points of developmental time.
- 42 brain specimens, male and female
- 13 developmental stages: post-conception week (pcw) 5-7 to 42 years old
RNA Sequencing is like reading a cell's mind¶



Load the data into flotilla via embark¶
%matplotlib inline
import flotilla
study = flotilla.embark(flotilla._brainspan)
A look behind the magic¶
flotilla._brainspan is just a link to a JSON file:
flotilla._brainspan
This json follows the datapackage specification as outlined by the Open Knowledge foundation.
! curl https://s3-us-west-2.amazonaws.com/flotilla/brainspan_batch_corrected_for_amazon_s3/datapackage.json
Back to the hypothesis: The human brain has different genes expressed in different regions¶
To address the question of how the expression varies across regions, we will Principal component analysis.
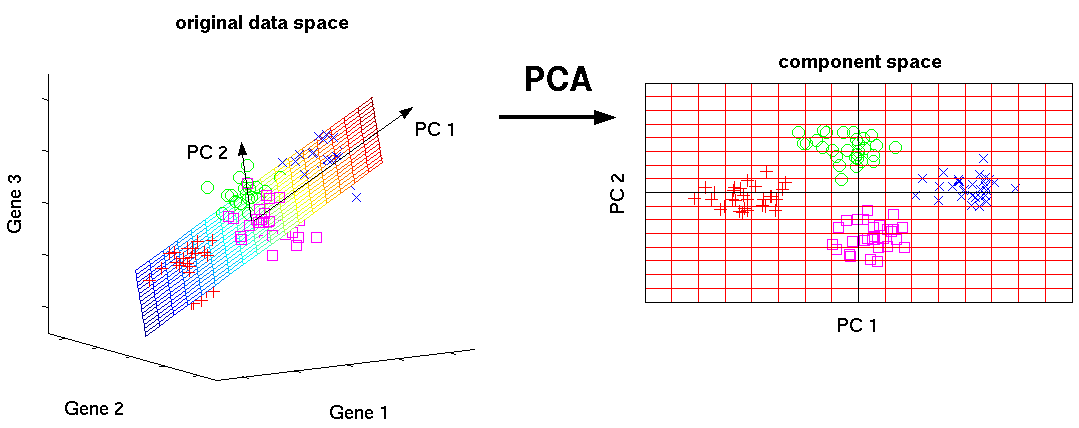
Principal Component Analysis (PCA)¶
Principal Component Analysis (PCA) is a dimensionality reduction algorithm which transforms a high-dimensional space like gene expression, to smaller dimensions, like just two for x- y- plotting.
# custom list: https://www.dropbox.com/s/wyjak3oh6z5myfm/cell_cycle_genes.txt?dl=0
study.interactive_pca()
Hypothesis: Cells in the hippocampus use genes unique to its function¶
The hippocampus is involved in memory, and we hypothesize that these cells have a unique molecular profile (set of genes that are expresssed). To accomplish this, we will use a classifier on our data to identify genes which separate hippocampal samples from non-hippocampal samples.
By default, flotilla uses an "Extremely Randomised Trees" Classifier (ExtraTreesClassifier), which takes random subsets of the data many times to create decision trees, like this one for deciding whether to play outside:

study.interactive_classifier()
Cilia are important for memory development¶
FOXJ1, C1orf88, TEKT1 are all involved in development of cilia, fingerlike protrusions from cells. Development of these cilia has been show to be important in memory formation.
So it looks like our classifier picked up the right things!



Acknowledgements¶
- Gene Yeo and the Yeo Lab, especially:
- Michael Lovci
- Yan Song
- Boyko Kakaradov
- Patrick Liu
- Leen Jamal
- Gabriel Pratt

Funding:
